Se publica un seguimiento en pacientes relocalizados por contacto estrecho en China infectados con la variante de preocupación Delta.
Infección viral y transmisión en un gran brote bien rastreado causado por la variante Delta SARS-CoV-2 Jin Lu. Publicado el 20 de Agosto.
Las medidas de control de la enfermedad, incluida la frecuencia de las pruebas de población, la cuarentena en fase presintomática y la mejora de la vigilancia genética, deben ajustarse para tener en cuenta la creciente prevalencia de la variante Delta a nivel mundial.
Menos tiempo presintomático 3,7 días de incubación contra 6 de la variante anterior . (Cuánto valor tiene un PCR para viajar).
Tiene mil veces carga viral que las otras cepas. Necesita más anticuerpos en menos tiempo. Por eso requiere dos o tres dosis, seis a ocho meses de cobertura.
Estos datos ponen de relieve que la variante Delta podría ser más infecciosa durante la primera etapa de la infección
Transmisión intergeneracional más rápida. Niños a adultos.
Se informa de la primera transmisión local de la variante Delta SARS-CoV-2 en China continental. Las 167 infecciones se podrían remontar al primer caso del índice. La investigación sobre las pruebas pcr secuenciales diarias de los sujetos en cuarentena relocalizados por contacto indicó que la carga viral de la primera prueba positiva de infecciones delta fue ~ 1000 veces mayor que la de las infecciones por cepas 19A / 19B en la ola epidémica inicial de 2020, lo que sugiere la tasa de replicación viral más rápida potencial y más infecciosidad de la variante Delta en la etapa temprana de la infección. Los 126 datos de secuenciación de alta calidad y los datos epidemiológicos fiables indicaron que algunas variantes menores de nucleótido único (iSNVs) dentro del huésped podían transmitirse entre los huéspedes y finalmente fijarse en la población del virus durante el brote. La transmisión menor de iSNVs entre donante-receptor contribuye con al menos 4 de las 31 sustituciones identificadas en el brote, lo que sugiere que algunos iSNVs podrían surgir rápidamente y alcanzar la fijación cuando el virus se propague rápidamente.
Las medidas de control de la enfermedad, incluida la frecuencia de las pruebas de población, la cuarentena en fase presintomática y la mejora de la vigilancia genética, deben ajustarse para tener en cuenta la creciente prevalencia de la variante Delta a nivel mundial.
Durante la propagación mundial del SARS-CoV-2, surgieron las variantes genéticas de los virus, y se ha demostrado que algunas son más transmisibles o que podrían escapar de la inmunidad del huésped, lo que planteaba un mayor riesgo para la salud pública mundial 1–3.
Un linaje genético emergente, B.1.617, ha sido dominante en el mayor brote de COVID-19 en India desde marzo de 2021, ganando la atención mundial. Un sublineado, B.1.617.2, con mutaciones de la proteína spike L452R, T478K y P681R, representa ~ 28% casos secuenciados en la India y reemplazó rápidamente a otros linajes para llegar a ser dominantes en múltiples regiones y países (https://outbreak.info/ 186)4.
El B.1.617.2 ha sido etiquetado como Variante de Preocupación (VOC), Delta (https://www.who.int/activities/tracking-SARS-CoV-2-variants 343). El perfil virológico de este VOC es necesario ser ilustrado urgente.
El 21 de mayo de 2021 se identificó la primera infección local de la variante Delta en China continental.
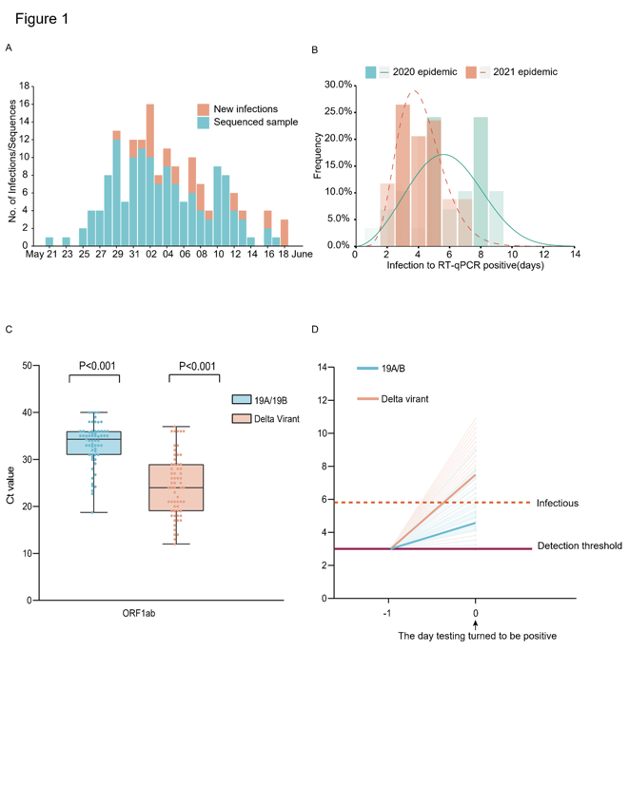
Al igual que en la primera epidemia de enero de 2020, se han llevado a cabo intervenciones estrictas que incluyen pruebas de detección de población, rastreo de contactos activados y cuarentena/aislamiento centralizado.
Sin embargo, a diferencia de las transmisiones restringidas en 2020, se ha observado una transmisión intergeneracional sucesiva en la epidemia de 2021. Aquí, investigamos los datos epidemiológicos, genéticos, y serológicos de este brote bien-trazado para caracterizar el perfil virológico de la variante del Delta SARS-CoV-2 y discutir cómo las estrategias de la intervención necesitan ser mejoradas en la carrera contra esta variante emergente.
Resultados
Las cargas virales en las infecciones Delta fueron ~ 1000 veces más altas que las de las infecciones por cepas 19A / 19B anteriores en el día en que se detectaron los virus por primera vez
Desde el primer caso índice identificado el 21 de mayo de 2021 hasta el último caso reportado el 18 de junio de 2021, se identificaron un total de 167 infecciones locales (Figura 1A). Todos estos casos se podían remontar epidemiológico o genético de nuevo al primer caso del índice. Una característica epidemiológica notable de la variante Delta es el intervalo serial más corto en comparación con las cepas tempranas de Wuhan u otras variantes de COV6-8. Sin embargo, los parámetros críticos antes de la aparición de la enfermedad siguen siendo esquivos, incluyendo cuándo los virus se pueden detectar en un sujeto después de la exposición y qué tan infecciosos son. Aquí, investigamos los datos de los sujetos en cuarentena en este brote y los comparamos con la epidemia anterior de 2020 causada por cepas genéticas 19A/19B.
Los sujetos centrales en cuarentena son los contactos estrechos de los casos confirmados/infecciones asintomáticas. Una vez que una nueva infección fue identificada, sus contactos cercanos fueron trazados inmediatamente, aislado centralmente y la prueba diaria de la polimerización en cadena fue realizada.
El conjunto de datos de sujetos en cuarentena nos permitió determinar el intervalo de tiempo entre la exposición y el alcance de la carga viral detectable por PCR en los sujetos infectados. Teniendo en cuenta que el tiempo de exposición exacto de las transmisiones intrafamiliares era difícil de identificar, deducimos los pares de transmisión intrafamiliar de nuestro análisis de intervalo de tiempo. Nuestros resultados mostraron que el intervalo de tiempo desde la exposición al primer PCR positivo en la población en cuarentena (n=29) fue de 6,00 (IQR 5,00-8,00) días en la epidemia de 2020 (pico a los 5,61 días) y fue de 4,00 (IQR 3,00-5,00) días en la epidemia de 2021 (n=34) (pico a los 3,71 días) (Figura 1B).
A continuación, evaluamos las cargas virales relativas cuando los virus SAS-CoV-2 se detectaron por primera vez en los huéspedes. Comparado a las tensiones 19A/19 B, las cargas virales relativas en las infecciones de la variante delta (62 casos, valor del Ct 24,00 (IQR 19,00~29,00) para el gene de ORF1ab) eran 1260 veces más altas que las infecciones de las tensiones 19A/19B (63 casos, valor del Ct 34,31 (IQR 31,00~36,00) para el gen de ORF1ab) en el día en que los virus primero fueron detectados (cuadro 1C). Considerando las pruebas diarias realizadas para los sujetos aislados centrales desde el inicio de la cuarentena, se propuso la mayor tasa de crecimiento dentro del huésped de la variante Delta, lo que llevó a las mayores cargas virales en los puntos de tiempo en que los nucleótidos virales superan el umbral de detección por PCR (Figura 1D). De forma similar al estudio realizado por Roman et.al9., encontramos muestras con valor de Ct superior a 30 (<6×105 copias/mL de virus) nunca producen un aislado infeccioso in vitro. Para las infecciones de la variante Delta, el 80,65% de las muestras contenían >6×105 copias/mL en hisopos orofaríngeos cuando se detectaron los virus en primer lugar en comparación con las muestras del 19,05% en las infecciones 19A/19B. Estos datos ponen de relieve que la variante Delta podría ser más infecciosa durante la primera etapa de la infección (Figura 1D).
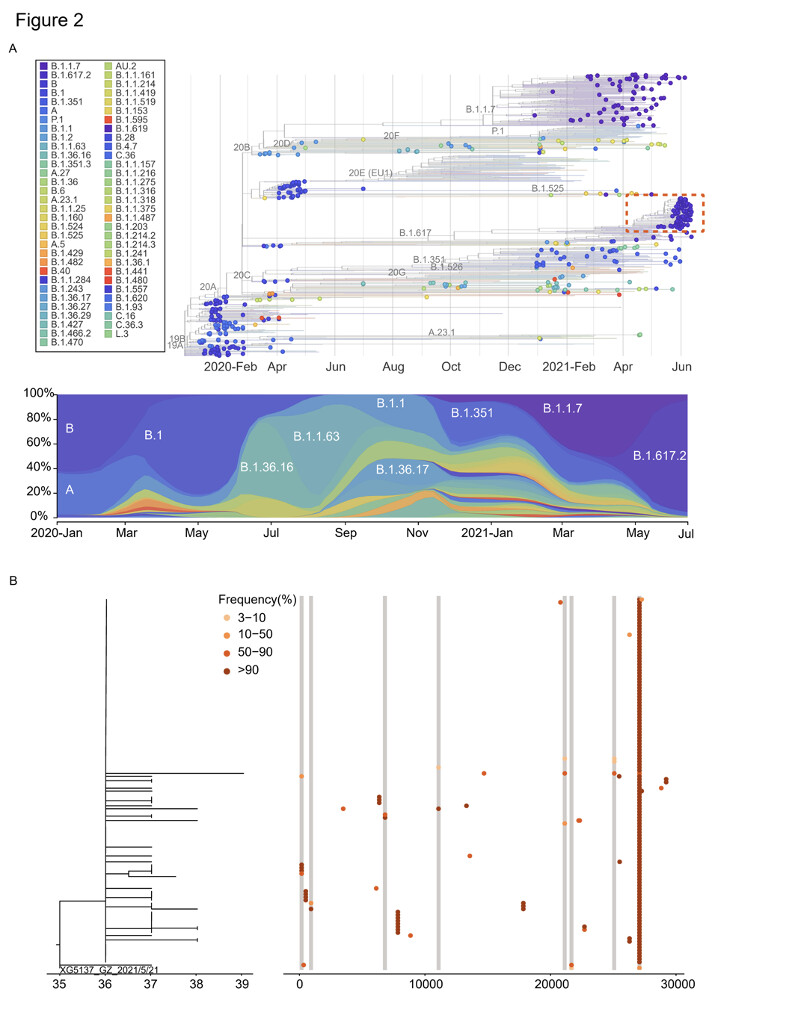
Figura 1: Resumen de la epidemiología y detección precoz de la variante Delta SARS-CoV-2 en Guangdong. (A) Las series temporales de 167 infecciones confirmadas por laboratorio se originaron a partir del primer caso índice el 21 de mayo. Los números diarios de nuevas infecciones mostradas en rojo y muestras con secuencias de alta calidad (cobertura>95%) se muestran en azul. (B) Estimar el marco de tiempo entre la exposición al primer positivo de RT-PCR en sujetos en cuarentena. Las distribuciones más ajustadas de la exposición al intervalo positivo RT-qPCR para las infecciones causadas por la variante Delta (n=34) y las cepas 19A/19B (n=29). Las líneas representan las distribuciones más ajustadas del intervalo. Las barras representan el número acumulado del intervalo (días). (C) Valores de Ct de las infecciones de la variante Delta (n =62) y las infecciones anteriores de las cepas 19A/19B (n = 63) en sujetos en cuarentena cuando la prueba de detección se vuelve por primera vez positiva. Los puntos representan la distribución del valor del Ct probada por RT-PCR en el gen ORF1ab (izquierda) y el gen N (derecha). Las gráficas de caja indican la mediana y el rango intercuartílico (IQR); los bigotes representan los valores máximo y mínimo. (D) Esquema de la relación entre la tasa de crecimiento viral y las cargas virales relativas en el día en que se detectaron por primera vez los virus (Día 0). La carga viral de las tensiones 19A/19B y de las infecciones de la variante del delta el día 0 eran calculadas. La línea discontinua horizontal en púrpura representa el umbral de detección de la prueba RT-PCR; la línea discontinua en rojo representa el límite inferior por encima del cual los virus infecciosos podrían ser potencialmente aislados.
Como sabemos, los individuos experimentan un período latente después de la infección, durante el cual los títulos virales son demasiado bajos para ser detectados. A medida que la proliferación viral continúa dentro del huésped, la carga viral eventualmente alcanzará un nivel detectable y se volverá infecciosa. Saber cuándo una persona infectada puede propagar virus es esencial para diseñar estrategias de intervención para romper las cadenas de transmisión. Sin embargo, esto es difícil de estudiar con base en investigaciones clínicas ya que más del 50% de la transmisión ocurrió durante la fase presintomática10. Nuestra investigación en los sujetos en cuarentena sugirió para la variante Delta, la ventana de tiempo desde la exposición a la detección de virus fue picos en ~ 3,7 días y presentó un mayor riesgo de infecciosidad / transmisión cuando el virus fue detectado por primera vez. En respuesta a este notable parámetro viral, el gobierno exigió a las personas que salen de la ciudad de Guangzhou desde aeropuertos, estaciones de tren y estaciones de autobuses de enlace que muestren pruebas de una prueba negativa de COVID-19 dentro de las 72 horas el 6 de junio y se acorten aún más a 48 horas el 7 de junio, en contraste con los siete días de la epidemia de 2020.imagen630×804 82 KB
iSNVs menores transmitidos entre huéspedes y fijos en la población de virus
Las intervenciones no farmacéuticas en Guangdong se centran principalmente en la investigación epidemiológica, el rastreo de contactos y las pruebas masivas. Se han realizado aproximadamente 30 millones de pruebas PCR desde el 26 de mayo de 2021 hasta el 8 de junio de 2021. Las pruebas y el cribado intensos de la población de alto riesgo hacen que las transmisiones crípticas sean poco probables y todas las infecciones identificadas podrían estar conectadas directas (por contacto directo) o indirectamente (permaneciendo o visitando la misma área). Además, todas las secuencias se podían remontar genético de nuevo al primer caso del índice. Esto nos brindó una oportunidad única para caracterizar la transmisión viral a una escala más fina, particularmente el alcance de la diversidad genética de virus transmitida entre los huéspedes. La secuenciación profunda del genoma completo se realizó en todas las infecciones identificadas mediante el uso del juego de cebadores Artic con la plataforma Illumina, y se obtuvieron 126 genomas virales de alta calidad (cobertura>95%), cubriendo el 75% de todas las infecciones identificadas (Figura 1A).
Análisis filogenético realizado mediante la inclusión de 346 secuencias de casos importados que viajaron desde 66 países a Guangdong entre marzo de 2020 y junio de 2021. Para las secuencias de referencia, se seleccionaron aleatoriamente 50 secuencias de genomas de cada clado definido (13 clados) en función de la clasificación nextstrain (https://nextstrain.org/ 77) y VOC notificado (Alfa, Beta, Gamma, Delta).
La dinámica de la distribución del linaje viral en estos casos importados reveló aproximadamente la circulación de linajes genéticos del SARS-CoV-2 a nivel mundial y también destacó los desafíos para el control y la prevención de enfermedades en Guangdong, China (Figura 2A).
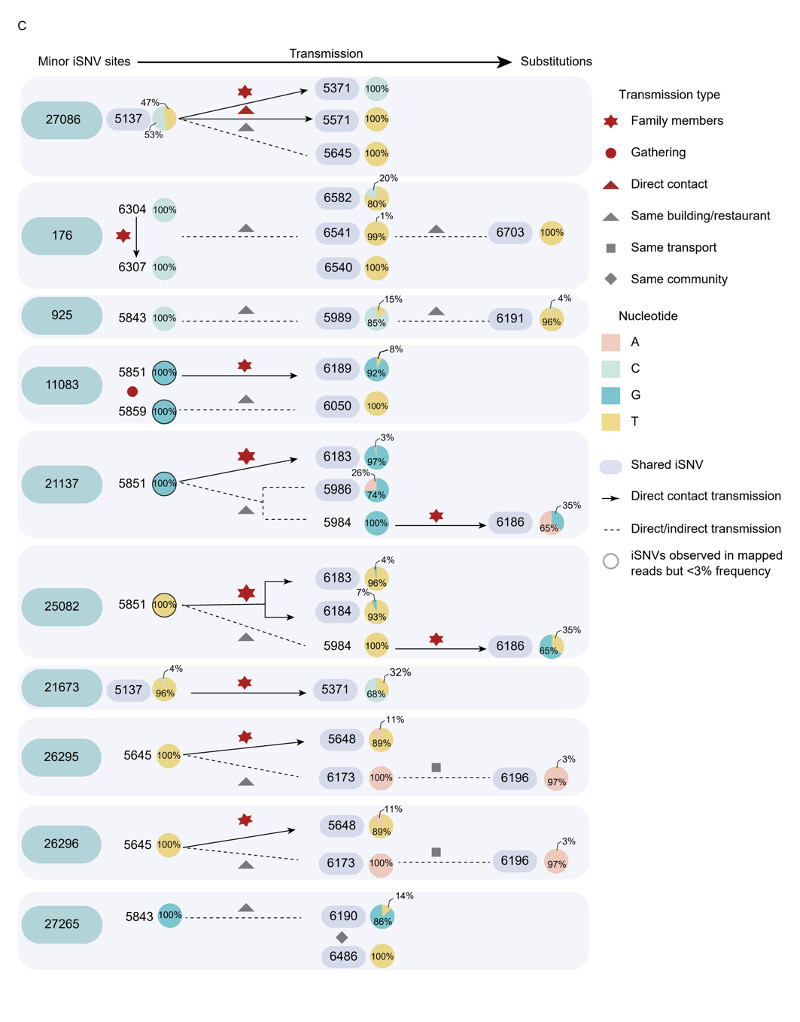
Figura 2: Filogenias virales y dinámica de transmisión del brote de Guangzhou. A)
El árbol filogenético resuelto en el tiempo se construyó con la tubería nextstrain mediante la inclusión de secuencias seleccionadas aleatoriamente de diferentes linajes genéticos, COV y secuencias de Guangdong recogidas de infecciones locales y casos importados, enero de 2020 – junio de 2021. Las secuencias del brote de la variante Delta entre el 21 de mayo de 2021 y el 18 de junio de 2021 se resaltaron con una caja roja. La dinámica de los linajes del SARS-CoV-2 identificados en Guangdong se mostró en el árbol de máxima verosimilitud del panel inferior (B) de 126 secuencias muestreadas del brote de Guangzhou. La frecuencia de SNV (%) también fue marcada con los puntos coloreados que colindaron con el árbol. (C) Relación de transmisión entre secuencias con los iSNVs menores en (B) y secuencias que tienen estas varianzas fijas (frecuencia alter >50%). El gráfico circular representaba la frecuencia de los iSNVs. La flecha mostró la transmisión directa del contacto con alta confianza. La línea del guión indicó que la transmisión directa o indirecta puede ocurrir entre dos secuencias muestreadas.
Las filogenias virales del brote de Guangzhou se construyeron en base a la secuencia de consenso de cada muestra, con ramas que indicaban el número de mutaciones en la secuencia de consenso entre las muestras y el eje x indicaba las mutaciones totales de la secuencia de referencia (Wuhan-Hu-1, MN908947). La secuencia de consenso para cada muestra se generó con base en la varianza mayoritaria (>50%) en cada posición. Todas las secuencias de brotes de Guangzhou segregadas en un solo grupo (Figura 2A). En comparación con el primer caso índice (XG5137_GZ_2021/5/21), se identificaron 31 sustituciones de 126 casos durante el brote de 26 días (Figura 2B). La secuencia más distante presentó sólo cuatro nucleótidos de diferencia de la muestra de caso índice. Esto sugiere que, durante un brote, la tasa de sustitución relativamente baja del SARS-CoV-2 presenta desafíos para inferir las cadenas de transmisión puramente basadas en secuencias de consenso11-13. Para inferir las variaciones menores de la secuencia junto con la propagación viral, estimamos la diversidad de virus dentro del huésped para cada muestra mediante el mapeo de sitios polimórficos en el genoma de consenso del caso índice (XG5137_GZ_2021/5/21) para generar una lista de variantes de nucleótido único intra-huésped (iSNVs). Los iSNVs menores fueron llamados estableciendo el 3% como el umbral de la frecuencia de alelos menores para excluir los posibles errores de PCR y secuenciación 12,14,15. Hubo secuencias con variantes menores de nucleótido único intra-huésped (iSNVs) en 10 de 31 posiciones sustituidas que pueden arrojar luces sobre cómo las variantes han surgido, crecido y finalmente arreglado durante la epidemia. Enumeramos las secuencias con estos iSNVs menores y las secuencias tienen las variantes correspondientes fijas (Figura 2B). La pregunta central que nos gustaría responder es que las variaciones genéticas acumuladas observadas en la epidemia de SARS-CoV-2 fueron dominantes por mutaciones de novo en un individuo o la transmisión de iSNVs seguidas de la fijación en el nuevo huésped. El rastreo de contactos y la investigación epidemiológica nos permitieron asignar la relación epidemiológica de estas secuencias con una alta confianza. Como se muestra en la Figura 2C, al menos algunos iSNV menores podrían transmitirse con éxito del donante al receptor o receptores. Los iSNVs menores C27086T podrían transmitirse desde el caso de índice a 2 de los 3 destinatarios. Con la propagación de los virus, esta sustitución se fijó en la población de virus en el brote (Figura 2B). Las sustituciones C925T, T21673C y G27265T encontradas en los casos 6191, 5371 y 6486 podrían tener los iSNVs correspondientes rastreados hasta los posibles donantes (Figura 3C). Sin embargo, también observamos que algunos iSNVs menores incluyen G11083T en las muestras 5851 y 5859, G21137A y T25082G en la muestra 5851 que no pudieron ser llamados (umbral como 3%) debido a la baja frecuencia pero pudieron ser observados en las lecturas mapeadas al genoma de referencia (Figura S1). El muestreo y la secuenciación de alta densidad indican que la variación genética generada durante la epidemia se debió, al menos en parte, a la transmisión de iSNVs entre donante-receptor, aunque las sucesivas transmisiones de iSNVs no se observaron posiblemente debido a las intervenciones sobre la transmisión del virus. Más importante aún, esta transmisión pudo observarse repetidamente cuando un donante tenía múltiples receptores (Figura 2C, sitio 27086). En esta circunstancia, es más probable que surjan algunos iSNV sars-cov-2 que mejoran la transmisión o que escapan inmunemente cuando el virus se propaga rápidamente.imagen796×1016 134 KB
imagen800×1022 125 KB
En este estudio, caracterizamos una gran cadena de transmisión originada a partir de la primera infección local de la variante delta sars-cov-2 en China continental. Se propone una tasa de replicación viral potencial más alta de la variante Delta, lo que lleva a las cargas virales en las infecciones Delta a ser ~ 1000 veces más altas que las infecciones por cepas 19A / 19B en el día en que las pruebas dan positivo. Esto pone de relieve que es muy probable que la infecciosidad de la variante Delta durante la etapa temprana de la infección, y la frecuencia del cribado poblacional debe optimizarse para la intervención16. La mayor infecciosidad de las infecciones de la variante Delta en fase presintomática pone de relieve la necesidad de una cuarentena oportuna para los casos de infección sospechosos o contactos estrechos antes del inicio clínico o el cribado por PCR.
Aunque los SNV intrahospitalario se encuentran en un nivel bajo, la transmisión menor del iSNV se observa resultando en una parte de las sustituciones fijas en la población del virus durante el brote.
Estos datos indican que alguna ventaja o mutaciones neutras, incluso a baja frecuencia, podrían aumentar y fijarse en la única generación de transmisión, y alcanzar aún más la población de virus si la epidemia no pudiera contenerse bien.
