Henry Grabowski , Joseph A. DiMasi , y Genia Long

La Ley de Reducción de la Inflación (IRA, por sus siglas en inglés) de 2022 afectará los incentivos para la inversión biofarmacéutica al reducir los precios promedio esperados y la rentabilidad. La Sección 1192 de la IRA crea un nuevo Programa de Negociación de Precios de Medicamentos (DPNP, por sus siglas en inglés) con Precios Máximos Justos de Medicare para medicamentos seleccionados. Los Precios Máximos Justos reales reflejarán las determinaciones de los Centros de Servicios de Medicare y Medicaid (CMS, por sus siglas en inglés) en función de ciertos factores específicos del producto y están sujetos a topes (pero no mínimos) exigidos por la legislación. Los CMS anunciaron los Precios Máximos Justos para los primeros diez medicamentos en agosto de 2024; entrarán en vigencia en enero de 2026. Reflejaron grandes descuentos porcentuales sobre los precios de lista y ahorros netos generales significativos en relación con el gasto de Medicare de 2023, teniendo en cuenta los reembolsos y ciertos otros pagos. 1 Aunque la IRA no le da a CMS autoridad sobre los precios para todas las personas aseguradas, los Precios Máximos Justos se harán públicos y, dadas las presiones políticas y la dinámica del mercado, pueden afectar los precios para otros pagadores, como las juntas estatales de asequibilidad de medicamentos recetados autorizadas para establecer límites de pago superiores. Además, existe presión para aplicar el DPNP antes y a más medicamentos.
Los medicamentos que no se enfrentan a la competencia de genéricos o biosimilares pueden ser seleccionados para el DPNP siete años después de la aprobación inicial en los EE. UU. si se aprueban según la Ley Federal de Alimentos, Medicamentos y Cosméticos (“medicamentos de moléculas pequeñas”) u once años si se autorizan según la Ley del Servicio de Salud Pública (“biológicos”). Los Precios Máximos Justos entran en vigencia dos años después (nueve años después de la aprobación en los EE. UU. para las moléculas pequeñas, trece para los biológicos).
Como resultado de los plazos del DPNP, la ley introduce nuevas consideraciones en las decisiones de inversión de los fabricantes posteriores a la aprobación. Los Precios Máximos Justos entrarán en vigor y reducirán los ingresos antes del final previsto del período de exclusividad en el mercado (el tiempo entre el lanzamiento en Estados Unidos y la entrada de los equivalentes genéricos, que ha oscilado entre un promedio de 12,2 años y 14,6 años para los medicamentos de moléculas pequeñas desde 1995-96). 2 Como resultado, las inversiones en indicaciones posteriores a la aprobación se volverán menos atractivas económicamente en relación con las alternativas. Además, como las fechas de elegibilidad del DPNP son cuatro años anteriores para las moléculas pequeñas que para los productos biológicos, los incentivos relativos para la inversión posterior a la aprobación serán diferentes.
El debate sobre los efectos del DPNP se ha centrado en evaluaciones muy diferentes del impacto en el número de nuevos medicamentos introducidos. 3 , 4 Sin embargo, el impacto potencial en la innovación posterior a la aprobación y las indicaciones adicionales se ha pasado por alto en gran medida. Aunque la selección de medicamentos se realiza en forma gradual, las decisiones de inversión posteriores a la aprobación se verán afectadas antes, ya que los fabricantes e inversores reevalúan sus carteras y actualizan los supuestos de planificación. Aunque la Oficina de Presupuesto del Congreso (CBO) no incluyó la dinámica posterior a la aprobación en sus estimaciones del impacto presupuestario, 3 más tarde destacó la necesidad de investigar el impacto en las decisiones de las empresas sobre la inversión en indicaciones posteriores a la aprobación. 5 Una descripción del panorama anterior a la IRA de la actividad posterior a la aprobación, como la que proporcionamos aquí, establece una base para futuras evaluaciones de los impactos de la IRA en la innovación de medicamentos posterior a la aprobación.
Nos centramos en los medicamentos oncológicos por varias razones. En primer lugar, están sujetos a incentivos legislativos cuyo valor se ve disminuido por la IRA, incluidos los incentivos de exclusividad para medicamentos huérfanos, pediátricos y de nueva investigación clínica destinados a fomentar la inversión. Las indicaciones múltiples son características importantes del desarrollo de medicamentos oncológicos, que reflejan vías de enfermedad compartidas. 6 Con el tiempo, los medicamentos oncológicos pueden probarse en diferentes tipos de tumores, en múltiples líneas de terapia (uso de línea posterior, de línea temprana), para su uso en combinación con diferentes agentes y como monoterapia, y en diferentes poblaciones de pacientes. Además, a menudo se aprueban para indicaciones huérfanas. Aunque los medicamentos con una designación huérfana están exentos de los Precios Justos Máximos, las designaciones o indicaciones huérfanas posteriores eliminan esta exención, aunque pueden aplicarse otras.
En segundo lugar, algunas partes interesadas han destacado la oncología como un área de preocupación en el marco de la IRA. 7 Los medicamentos oncológicos representaron 54.000 millones de dólares en gastos combinados de las Partes B y D de Medicare en 2020. 8 Dado que se estima que los costos del cáncer de las Partes A, B y D aumentarán un 34 por ciento entre 2015 y 2030, alcanzando los 246.000 millones de dólares en dólares de 2019, la oncología seguirá siendo importante para el gasto de Medicare. 9 Además, con los avances continuos en la detección, la detección temprana y el tratamiento, y la supervivencia del cáncer, desarrollar todo el potencial de los medicamentos es clínica y económicamente importante. 10 Por último, los medicamentos oncológicos incluyen tanto moléculas pequeñas como productos biológicos, por lo que se aplican ambos plazos de Precio Justo Máximo.
Porcentaje acumulado de ensayos posteriores a la aprobación, por años desde la aprobación original del fármaco en EE. UU. hasta el final del ensayo, para fármacos oncológicos aprobados por primera vez en EE. UU. durante 2000-2
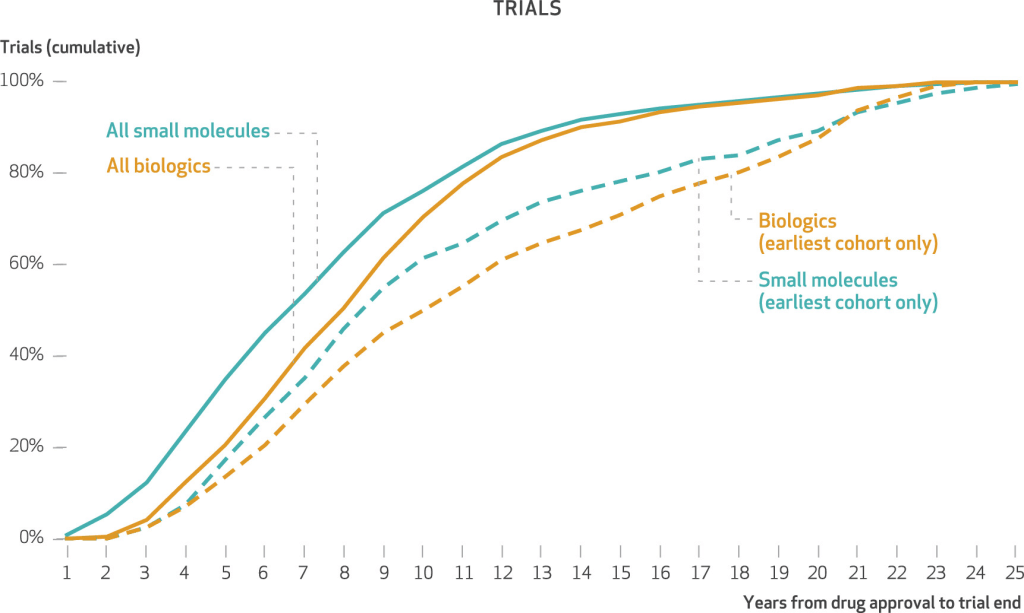
Cálculos de los autores de las fechas de finalización de los ensayos primarios notificadas a partir de la base de datos en línea ClinicalTrials.gov (véase la nota
16 en el texto), buscando en el campo intervención/tratamiento por nombre genérico del fármaco ensayos intervencionistas financiados por el fabricante para fármacos oncológicos aprobados en los EE. UU. durante el período 2000-21. En el apéndice se describen otras restricciones aplicadas a los datos de los ensayos clínicos (véase la nota 12 en el texto). NOTAS “Fin del ensayo” es la fecha de finalización primaria notificada. Las cohortes de fármacos del año de aprobación más recientes reflejan menos años de observación posterior a la aprobación y, por lo tanto, una mayor censura a la derecha de los datos y porcentajes acumulativos más altos en un año determinado. La cohorte del año de aprobación más temprana es 2000-04;
Porcentaje acumulado de indicaciones posteriores a la aprobación, por años desde la aprobación original del fármaco en EE. UU. hasta la aprobación de la indicación, para fármacos oncológicos aprobados por primera vez en EE. UU. durante 2000-21
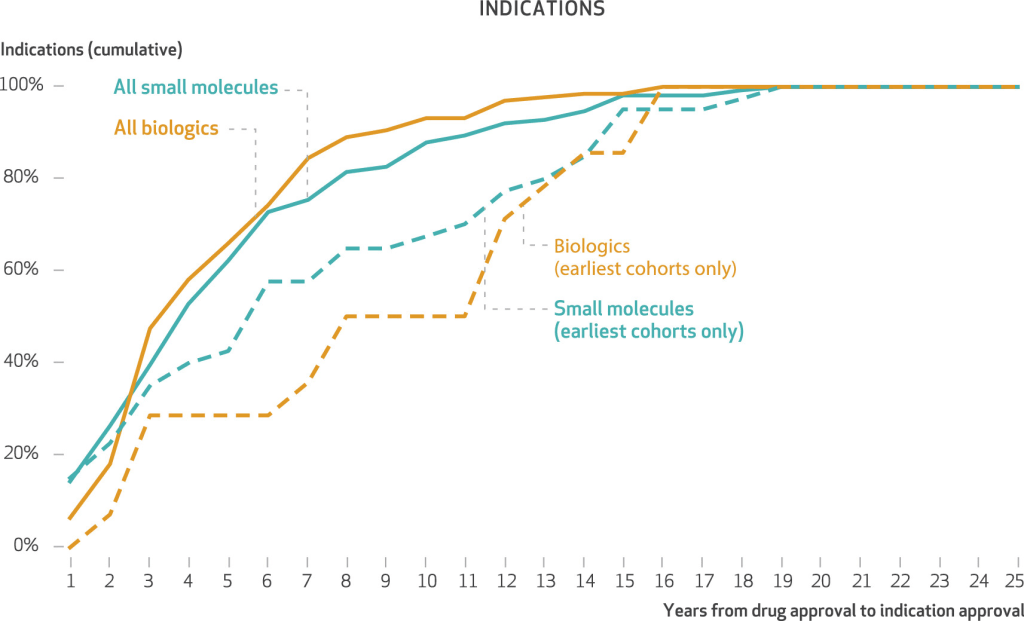
Cálculos de los autores de las fechas de acción en las etiquetas que se encuentran en Drugs@FDA (ver nota
11 en el texto), buscando por nombre del fármaco, para fármacos oncológicos aprobados en los EE. UU. durante el período 2000-21. NOTAS Las cohortes de fármacos de año de aprobación más recientes reflejan menos años de observación posterior a la aprobación y, por lo tanto, más censura a la derecha de los datos y porcentajes acumulados más altos en un año determinado. Las indicaciones aprobadas reflejan menos puntos de datos totales; las dos primeras cohortes de año de aprobación son 2000-04 y 2005-09.
Discusión
En el caso de los fármacos oncológicos aprobados entre 2000 y 2021, descubrimos que más de la mitad de todas las indicaciones y aproximadamente dos tercios de los ensayos clínicos financiados por la industria se produjeron después de la aprobación. Otros han encontrado resultados similares: en el caso de los fármacos aprobados entre 1995 y 2019 (en todas las áreas de enfermedad), las indicaciones posteriores a la aprobación fueron aproximadamente el 40 por ciento de todas las indicaciones. 18 , 19 Nuestro estudio proporcionó hallazgos complementarios: ampliamos el período de estudio hasta 2021, comparamos fármacos de moléculas pequeñas y biológicos, proporcionamos análisis paralelos de ensayos clínicos y examinamos características adicionales de los fármacos y las indicaciones.
Los medicamentos oncológicos suelen seguir un camino de desarrollo que anticipa indicaciones adicionales posteriores a la aprobación, y los fabricantes priorizan las indicaciones que tienen el camino más rápido hacia la aprobación, generalmente para una población de pacientes más reducida. En un estudio de 2022 sobre veinticinco presentaciones oncológicas en los EE. UU. y Europa, otros descubrieron que los medicamentos oncológicos generalmente se aprobaban primero para cánceres más raros con mayores ganancias de años de vida ajustados por calidad atribuidas, seguidos de indicaciones de mayor prevalencia. 6 Descubrimos que el camino posterior a la aprobación variaba, en algunos casos de un tipo de tumor a otro con un mecanismo biológico subyacente común (por ejemplo, la inmunoterapia pembrolizumab tenía treinta y seis indicaciones aprobadas en dieciocho áreas de enfermedad diferentes en nuestra muestra). En otros casos, el desarrollo procedió de una aprobación de línea posterior en un tipo de tumor a una de línea anterior, o de una combinación a una monoterapia, o con terapias combinadas adicionales probadas. A veces pueden aplicarse múltiples aspectos, como en el caso del pembrolizumab.
Los fabricantes tienen en cuenta muchos factores a la hora de decidir si invertir en investigación y desarrollo posteriores a la aprobación, entre ellos, las necesidades médicas no satisfechas, las poblaciones de pacientes que se beneficiarían y factores económicos como el tiempo restante previsto hasta que se produzca la entrada de medicamentos genéricos y se desplome la cuota de mercado de los medicamentos de marca (o, en el caso de los productos biológicos, la entrada de biosimilares, donde los impactos están menos bien establecidos) y el coste, la duración y las probabilidades previstas de éxito de los ensayos clínicos en cuestión. Al reducir los precios previstos, el DPNP reducirá las ganancias previstas de la inversión en indicaciones posteriores a la aprobación.
Aunque el cálculo será diferente para cada fármaco, en igualdad de condiciones, el DPNP creará desincentivos para ciertos tipos de actividades de desarrollo de fármacos posteriores a la aprobación: para moléculas pequeñas versus productos biológicos (porque los Precios Máximos Justos entran en vigencia antes), para fármacos que atienden desproporcionadamente a personas mayores y discapacitadas (porque tienen una mayor proporción de gasto de Medicare y, por lo tanto, una mayor exposición a las fechas del DPNP en comparación con los fármacos para poblaciones más jóvenes), y para ciertos ensayos posteriores a la aprobación de mayor duración o que comienzan más tarde y que finalizarían después o cerca de las fechas del DPNP.
Todos los ensayos arrojan información que puede influir en la práctica, pero no todos darán lugar en última instancia a indicaciones adicionales aprobadas en la etiqueta. Esto se refleja en nuestra muestra: aunque el 50 por ciento de los medicamentos no tenían indicaciones adicionales posteriores a la aprobación (en el momento del análisis), un porcentaje mucho menor (11 por ciento) no mostró ningún inicio de ensayos clínicos patrocinados por el fabricante posteriores a la aprobación. Por este motivo, el seguimiento de los cambios en los ensayos clínicos posteriores a la aprobación puede proporcionar una «alerta temprana» de los impactos del DPNP.
Se desconoce el impacto clínico, económico y de salud pública de las indicaciones post-aprobación que podrían dejarse de lado, y presumiblemente variará según el área terapéutica, el tamaño de la población, las necesidades médicas no satisfechas y las terapias alternativas disponibles. Otros han descubierto que las indicaciones posteriores a la aprobación afectan el comportamiento de los prescriptores. Utilizando la proporción de prescripciones según la Clasificación Estadística Internacional de Enfermedades y Problemas Relacionados con la Salud, Décima Revisión, como medida del valor clínico reflejado en las decisiones de prescripción, Benjamin Berger y coautores investigaron las indicaciones posteriores a la aprobación de medicamentos aprobados durante 1995-2019. Encontraron que en todas las áreas terapéuticas, y específicamente para la oncología, los médicos juzgaron que las aprobaciones de indicaciones posteriores eran clínicamente importantes, como se refleja en un aumento del 40 por ciento en la proporción de prescripciones durante el tiempo de la aprobación original. Teniendo en cuenta el uso fuera de etiqueta previo a la aprobación, los autores descubrieron que fue la aprobación de la FDA, no el inicio de los ensayos clínicos, lo que fue responsable del aumento, y que el aumento de la proporción fue mayor cuando la nueva indicación se refiere a una nueva área de enfermedad. 18 El impacto clínico de los medicamentos oncológicos varía en términos de impacto en la supervivencia, la calidad de vida u otras métricas, y puede ser difícil de predecir. El estudio de 2022 realizado por Daniel Tobias Michaeli y colegas sobre veinticinco lanzamientos oncológicos encontró que las indicaciones originales ocuparon una posición algo más alta en los resultados de una escala de beneficios clínicos estandarizada en comparación con las indicaciones posteriores, pero los autores no encontraron una diferencia estadísticamente significativa entre las dos, controlando otras características de aprobación regulatoria, ensayo y tratamiento. 6
Los medicamentos oncológicos incluyen tanto moléculas pequeñas como productos biológicos, y en nuestro análisis observamos la “penalización por píldora” de las moléculas pequeñas20 . Otros han sugerido que diferentes cronogramas del DPNP “podrían resultar en favorecer a los productos biológicos aún más que a las moléculas pequeñas, influyendo en futuras decisiones relacionadas con la financiación de la biotecnología y las líneas de desarrollo de medicamentos”. 21 Esto es coherente con nuestro hallazgo de que el porcentaje de ensayos de moléculas pequeñas posteriores a la aprobación que terminaron después de las fechas de selección e implementación de medicamentos relevantes del DPNP fue más del doble que el porcentaje de los productos biológicos, aunque los tiempos esperados desde la aprobación del medicamento hasta el final del ensayo fueron más cortos para las moléculas pequeñas.
El nuevo tope de $2,000 que el beneficiario debe pagar de su bolsillo para la Parte D puede compensar esta penalización por las píldoras hasta cierto punto. Se desconoce la magnitud relativa y el efecto neto de las diversas disposiciones de la IRA, así como el nivel final de los Precios Máximos Justos, el grado y el ritmo de la repercusión en el mercado comercial y la respuesta de las aseguradoras a otras disposiciones de rediseño de los beneficios de la Parte D. En todas las áreas de enfermedad, la CBO estimó que el efecto descendente agregado sobre el gasto de la Parte D para 2031 a partir del efecto combinado de los Precios Máximos Justos y los nuevos reembolsos por inflación sería aproximadamente ocho veces mayor que el efecto ascendente debido a la eliminación del tope de los gastos de bolsillo (un aumento del 2 por ciento frente a una disminución del 16 por ciento). 22
En cuanto a las posibles respuestas al nuevo desincentivo a la inversión posterior a la aprobación, se puede esperar que las empresas reconsideren sus carteras, avanzando con proyectos con valores esperados más altos en comparación con los más marginales, para moderar los impactos adversos en la innovación. Aunque excluimos los ensayos observacionales de nuestro análisis, el reciente borrador de orientación de la FDA sobre evidencia del mundo real en las presentaciones regulatorias puede sugerir un papel mayor para los estudios de evidencia del mundo real en la planificación de la evidencia de los fabricantes como respuesta a los ciclos de vida acortados de los productos resultantes del DPNP. 23 Un mayor énfasis en las aprobaciones aceleradas posteriores a la aprobación, cuando sea posible, es otra respuesta potencial a los ciclos de vida restringidos. El cuarenta y ocho por ciento (o 74 de los 155 medicamentos en nuestra muestra) fueron aprobados originalmente a través de una aprobación acelerada.
Conclusiones políticas
El análisis sugiere varias conclusiones importantes para los responsables de las políticas. En primer lugar, si la historia sirve de guía, es probable que la IRA y sus disposiciones sobre el DPNP tengan importantes impactos en la innovación posterior a la aprobación, y centrarse únicamente en el número de nuevos medicamentos introducidos proporciona sólo una imagen parcial. Las indicaciones posteriores a la aprobación y los ensayos financiados por la industria son una parte sustancial de todas las indicaciones y ensayos para medicamentos oncológicos, y las fechas pertinentes suelen ocurrir después o cerca de las fechas de selección e implementación del DPNP, en particular para las moléculas pequeñas, lo que sugiere que enfrentarán desincentivos debido al DPNP.
En segundo lugar, el impacto en las moléculas pequeñas será mayor que el impacto en los productos biológicos debido a los plazos del DPNP. La exposición a las fechas de selección de medicamentos del programa es más del doble para las moléculas pequeñas que para los productos biológicos, ya que las fechas de IRA son posteriores, aunque las duraciones esperadas desde la aprobación del medicamento hasta el final del ensayo son más cortas. Por lo tanto, la mayor exposición a la elegibilidad del DPNP se puede atribuir en gran medida a la diferencia de cuatro años entre los “objetivos”.
En tercer lugar, surgen varias áreas de probable impacto. Aproximadamente la mitad de las indicaciones posteriores a la aprobación que examinamos se aprobaron en nuevas áreas de enfermedades, en las que investigaciones anteriores han encontrado que las nuevas indicaciones son particularmente importantes para las decisiones de prescripción. 18 Otras indicaciones posteriores a la aprobación reflejan nuevas condiciones de terapia combinada, nuevas recomendaciones de líneas de terapia, nueva aprobación pediátrica u otras definiciones de poblaciones de pacientes (incluidas poblaciones definidas por anomalías genéticas o los resultados de otras pruebas).
El impacto en las indicaciones huérfanas podría ser sustancial: aproximadamente tres cuartas partes de los medicamentos tenían al menos una indicación huérfana, y aproximadamente la mitad de ellos tenían más de una (eliminando una exención del DPNP). Los ejemplos incluyen indicaciones huérfanas para pacientes pediátricos (por ejemplo, tratamiento de pacientes pediátricos de dos años o más con linfoma de Hodgkin clásico de alto riesgo no tratado previamente en combinación con doxorrubicina, vincristina, etopósido, prednisona y ciclofosfamida).
En cuarto lugar, el DPNP representa un cambio sustancial en los incentivos legislativos para la innovación biofarmacéutica, que han otorgado períodos adicionales de exclusividad de diversos tipos en áreas de alta prioridad de salud pública (por ejemplo, exclusividad pediátrica de seis meses, exclusividad de siete años para medicamentos huérfanos y exclusividad de tres años para investigaciones con nuevos productos químicos). Esto puede conducir a efectos no deseados, ya que la introducción de Precios Máximos Justos un número fijo de años después de la aprobación de los medicamentos reduce sustancialmente el valor de dichos incentivos. Por ejemplo, seis meses de exclusividad pediátrica al final de las patentes y exclusividades existentes no tendrían ningún efecto en la fecha del Precio Máximo Justo, y el valor de las ventas desde la fecha de vigencia del Precio Máximo Justo hasta la entrada en el mercado de los genéricos se reduciría. Cuanto mayor sea la reducción en los precios promedio de los medicamentos de Medicare, menor será el valor del incentivo de exclusividad pediátrica. Esto podría ser un motivo de preocupación, dado que, si bien las indicaciones pediátricas son una pequeña parte de todas las indicaciones, una parte sustancial de ellas se producen después de la aprobación.
Aunque la magnitud es incierta, se espera que las disposiciones sobre precios del DPNP creen desincentivos para los ensayos clínicos y las indicaciones posteriores a la aprobación. Dada la frecuencia, el momento y la importancia potencial de la actividad posterior a la aprobación en oncología, esto sugiere la importancia de monitorear las tendencias en relación con el nivel de referencia observado de innovación posterior a la aprobación para evaluar en qué medida la IRA desalienta la innovación posterior a la aprobación.
Entre las posibles respuestas de política se podría incluir que los CMS envíen una señal positiva a las futuras decisiones de inversión incorporando consideraciones de investigación y desarrollo posteriores a la aprobación en los Precios Máximos Justos. Aunque las directrices de los CMS establecen que las necesidades médicas no satisfechas se tendrán en cuenta en sus determinaciones de precios, entre otros factores, la ponderación de estos factores sigue sin estar clara. 24 En ausencia de esa acción administrativa, el valor de las inversiones en innovación posteriores a la aprobación y los incentivos legislativos previos basados en la exclusividad se verán reducidos.
