Caroline Horrow,1Sarah ME Gabriele, JD, LLM, MBE1S. Sean Tu, 1,2
JAMA 2024;184;(7):810-817. doi:10.1001/jamainternmed.2024.0836
La estructura de las múltiples patentes superpuestas, conocidas como marañas de patentes, para los medicamentos recetados más vendidos consiste en una compleja red de derechos de propiedad intelectual. Estas marañas se componen de numerosas patentes solicitadas y concedidas, que protegen no solo el principio activo del medicamento, sino también otros elementos como métodos de uso, procesos de fabricación, formulaciones y dispositivos de administración.
Pregunta central
¿Cuál es la estructura de las múltiples patentes superpuestas (es decir, marañas de patentes) para los medicamentos recetados más vendidos?
Hallazgos principales
En un estudio transversal que analizó 1429 patentes y solicitudes de patente asociadas a los diez medicamentos de marca con mayores ingresos en Estados Unidos en 2021, se observó que casi tres cuartas partes de dichas patentes se presentaron después de la aprobación por parte de la Administración de Alimentos y Medicamentos de EE. UU. (FDA). Las patentes posteriores a la aprobación, así como aquellas que protegen partes del medicamento distintas del ingrediente activo, contribuyeron decisivamente a la formación de una maraña patentaria densa y compleja.
Implicaciones y recomendaciones
Los resultados del estudio sugieren que, dado que la maraña de patentes de los medicamentos más vendidos está formada en gran medida por patentes solicitadas tras la aprobación de la FDA, se requiere un examen minucioso de estas solicitudes y de las patentes presentadas en esta etapa. Este análisis es fundamental para facilitar una competencia oportuna de medicamentos genéricos y biosimilares, lo que podría reducir los precios y ampliar el acceso para pacientes y pagadores.
Importancia del fenómeno
Los medicamentos de marca mantienen precios elevados en Estados Unidos durante los periodos de exclusividad de mercado protegidos por patentes. La existencia de múltiples patentes superpuestas puede retrasar durante años la entrada de competidores genéricos, prolongando así los altos precios y limitando el acceso a alternativas de menor costo.
Objetivo del estudio
El objetivo fue evaluar la composición de los entramados de patentes de los diez medicamentos recetados más vendidos en EE. UU. y comparar las características de las patentes presentadas durante el desarrollo del producto con aquellas solicitadas después de la aprobación de la FDA.
Diseño y contexto de la investigación
Este estudio transversal examinó la red de patentes estadounidenses que protegía a los diez medicamentos con receta de mayor facturación en 2021. La información se obtuvo de una base de datos pública de la Iniciativa para el Acceso a Medicamentos y el Conocimiento (IAM), analizando datos de patentes emitidas y solicitudes al 30 de junio de 2022. El análisis se realizó entre septiembre de 2022 y junio de 2023.
Principales resultados y medidas
El estudio se centró en la prevalencia de patentes presentadas antes y después de la aprobación de la FDA, los tipos de reivindicaciones presentes en las patentes concedidas (composición química, método de uso, proceso o síntesis, formulación y dispositivo de administración) y la densidad del entramado de patentes, medida por el número de patentes activas en un momento dado.
Resultados específicos
Los diez medicamentos analizados incluyeron cuatro de molécula pequeña y seis biológicos, vinculados a un total de 1429 patentes y solicitudes: 742 patentes emitidas (52%), 218 solicitudes pendientes (15%) y 469 solicitudes abandonadas (33%). Casi el 72% de las solicitudes de patente (1028) se presentaron después de la aprobación de la FDA, siendo esta proporción mayor para productos biológicos (80%) que para medicamentos de molécula pequeña (58%). La presentación de solicitudes de patente posteriormente a la aprobación alcanzó su punto máximo en los primeros cinco años para medicamentos de molécula pequeña y doce años para productos biológicos. De las 465 patentes emitidas tras la aprobación de la FDA, 189 (41%) contenían reivindicaciones de método de uso, 127 (27%) de formulación, 103 (22%) de proceso o síntesis, 86 (19%) de composición química y 46 (10%) de dispositivo. La densidad de patentes alcanzó su máximo trece años después de la aprobación de la FDA, con una mediana de 42 patentes activas por medicamento, el 66% de las cuales se presentaron tras la aprobación.
Conclusiones y relevancia
El estudio muestra que, entre los medicamentos recetados más vendidos en Estados Unidos, las patentes solicitadas tras la aprobación de la FDA y que cubren aspectos distintos al principio activo del fármaco son responsables de la formación de marañas de patentes. Para promover una competencia efectiva de genéricos y biosimilares, se requiere un análisis exhaustivo de estas solicitudes y patentes posteriores a la aprobación.
Introducción
Los elevados costos de los medicamentos de marca con receta que enfrentan los estadounidenses han sido una preocupación apremiante para los legisladores y los pacientes. 1 , 2 En Estados Unidos, los fabricantes de medicamentos de marca fijan precios altos durante los períodos de exclusividad de mercado, que pueden durar de 12 a 14 años o más, según las patentes que cubren el medicamento. 3 Una vez que expiran los períodos de exclusividad, la disponibilidad de medicamentos genéricos y biosimilares puede contribuir a precios sustancialmente más bajos y a una reducción del gasto para pacientes y aseguradoras. 4
Cada patente de medicamento suele otorgar 20 años de exclusividad de mercado. Las patentes se obtienen habitualmente para el principio activo de un medicamento; sin embargo, para los medicamentos de éxito, los fabricantes de fármacos de marca obtienen patentes adicionales sobre otros aspectos del fármaco, como su formulación, métodos de uso y procesos de fabricación. 5 , 6 Las múltiples patentes que protegen un medicamento de marca exitoso se conocen como marañas de patentes, y pueden retrasar y dificultar la disponibilidad de medicamentos genéricos y biosimilares, ya que los fabricantes de la competencia a menudo deben esperar a que expiren las patentes o presentar una demanda para que se invaliden ciertas patentes (o llegar a un acuerdo con el fabricante del medicamento de marca) antes de poder comercializar su producto. 7
La falta de competencia oportuna de genéricos y biosimilares en el mercado farmacéutico estadounidense, causada por la maraña de patentes, mantiene altos los costos de los medicamentos para pacientes y pagadores. Para abordar este problema, la administración Biden emitió una Orden Ejecutiva en julio de 2021 que instruye a la Administración de Alimentos y Medicamentos de los Estados Unidos (FDA) y a la Oficina de Patentes y Marcas de los Estados Unidos (USPTO) a cooperar «para ayudar a garantizar que el sistema de patentes, si bien incentiva la innovación, no retrase injustificadamente la competencia de medicamentos genéricos y biosimilares más allá de lo razonablemente contemplado por la ley aplicable». 8 Ambas agencias han acordado colaborar en soluciones políticas para «proteger contra la patentación de cambios incrementales y obvios a medicamentos existentes que no califican para patentes». 9 La FDA y la USPTO intercambiaron cartas que describen posibles iniciativas, como brindar a los examinadores de patentes acceso a más recursos de la FDA para investigar la aprobabilidad de las solicitudes y evitar otorgar patentes que no cumplan con los criterios legales básicos. 10 , 11 Hasta el momento, las agencias han comenzado a colaborar a través de una sesión conjunta de escucha pública y eventos de capacitación cruzada del personal, pero no han realizado cambios políticos importantes. 12
Para comprender mejor las vías para mejorar la competencia oportuna de genéricos y biosimilares, evaluamos las características de los entramados de patentes de los 10 medicamentos recetados más vendidos en EE. UU. en 2021. Comparamos las características de las patentes presentadas durante el desarrollo del producto con las patentes presentadas para estos productos después de la aprobación de la FDA.
Métodos
Fuentes de datos, extracción y codificación
Comenzamos con datos de patentes extraídos del Drug Patent Book, de acceso público, compilado por la Initiative for Medicines, Access, and Knowledge, una organización legal sin fines de lucro. 13 El Drug Patent Book se elaboró mediante la caracterización sistemática de los panoramas de patentes de los 10 medicamentos con los mayores ingresos netos por ventas en EE. UU. en 2021 (en este caso, los medicamentos incluían tanto fármacos de molécula pequeña como productos biológicos). La base de datos incluye información sobre patentes concedidas y solicitudes de patentes publicadas hasta junio de 2022. 14 La base de datos se construyó incluyendo patentes de los Productos Farmacéuticos Aprobados con Evaluaciones de Equivalencia Terapéutica (Libro Naranja) y la Base de Datos de Productos Biológicos Licenciados (Libro Púrpura) de la FDA (compilaciones disponibles públicamente de ciertos tipos de patentes que los fabricantes presentan a la FDA en el momento de la aprobación para todos los fármacos de molécula pequeña y en momentos posteriores para ciertos productos biológicos, respectivamente) listadas hasta junio de 2022 y complementada con patentes alegadas en litigios e identificadas a través de búsquedas en las bases de datos de propiedad intelectual Orbit Intelligence (Questel), CAS SciFinder (Sociedad Química Estadounidense), Lens.org y Espacenet. De acuerdo con la Regla Común, este estudio transversal estuvo exento de revisión, ya que se basó en datos disponibles públicamente y no involucró información personal de salud.
Para confirmar la relevancia de las patentes del Drug Patent Book para un medicamento en particular, realizamos una verificación cruzada con los listados históricos del Orange Book de 1985 a 2022, la base de datos de Unified Patents de litigios estadounidenses de 2000 a 2020 y la base de datos de Lex Machina de litigios estadounidenses de 2000 a junio de 2023. Utilizamos las fechas de vencimiento de las patentes que figuran en el Drug Patent Book. Para cualquier patente que figurara con un vencimiento superior a 20 años después de su fecha de presentación, realizamos una verificación cruzada con la lista pública de extensiones de plazo de patentes de la USPTO y el Centro de Patentes de la USPTO para confirmar si la patente había recibido un ajuste en su fecha de vencimiento.
Las fechas de aprobación de cada medicamento por la FDA y la autorización de equivalentes terapéuticos se identificaron a partir de Drugs@FDA. 15 Para facilitar la comparación entre medicamentos, convertimos las fechas de presentación, concesión y vencimiento de cada patente en el número de años antes o después de la aprobación de la FDA. Para evaluar las tendencias en las prácticas de presentación de los fabricantes, analizamos las fechas de presentación reales (las fechas en las que se presentaron las solicitudes a la USPTO) de todas las solicitudes de patente, incluidas las que dieron lugar a patentes emitidas y las que están pendientes o han sido abandonadas. El estado de cada patente o solicitud (activa, vencida, pendiente o abandonada) se determinó al 30 de junio de 2022, el final de la recopilación de datos. Para cada medicamento, calculamos el número y el porcentaje de solicitudes de patente y patentes emitidas presentadas por año antes y después de la aprobación de la FDA.
Características de la patente
Para nuestro análisis de las características de las patentes, solo se incluyeron las patentes emitidas, ya que contenían reivindicaciones que la USPTO había considerado patentables. Con base en las reivindicaciones presentes, las patentes se asignaron a una o más categorías: (1) reivindicaciones de composición química, que describen la entidad molecular; (2) reivindicaciones de método de uso, que describen el tratamiento o la prevención de una enfermedad, la evaluación de un diagnóstico o la alteración de un biomarcador asociado con la enfermedad; (3) reivindicaciones de proceso o síntesis relacionadas con la fabricación; (4) reivindicaciones de formulación, que describen una nueva forma farmacéutica, vía de administración, concentración o combinación de fármacos; y (5) reivindicaciones de dispositivo, que describen un mecanismo para administrar un producto en el cuerpo (eMethods en el Suplemento 1 ). Dado que la mayoría de las patentes contienen múltiples reivindicaciones, cada patente podría asignarse a múltiples categorías. Dos autores (CH y SMEG) codificaron de forma independiente muestras de 25 patentes y discutieron cada patente para llegar a un consenso sobre las definiciones de las categorías. Después de una revisión completa, las reivindicaciones poco claras fueron revisadas conjuntamente por los 2 codificadores y en discusión con otros autores según fuera necesario para llegar a un consenso. Comparamos el número y el porcentaje de patentes en cada categoría de reivindicación presentadas antes y después de la aprobación de la FDA.
Análisis estadístico
Para analizar la densidad de patentes, utilizamos las fechas de concesión y vencimiento para calcular el número de patentes activas en cada intervalo de un año, antes o después de la aprobación de la FDA. La densidad de patentes se definió como el número de patentes activas sobre un fármaco en un momento dado. Los datos se analizaron desde septiembre de 2022 hasta junio de 2023 utilizando Excel, versión 16 (Microsoft Corp).
Resultados
La muestra incluyó patentes para 4 fármacos de molécula pequeña y 6 productos biológicos ( Tabla ). Al 30 de junio de 2022, los fármacos habían sido aprobados durante 4,4 a 23,7 años (mediana [RIC], 10,1 [8,0-15,6] años). A estos 10 fármacos, vinculamos 1429 patentes y solicitudes de patente: 742 (52%) patentes emitidas, 218 (15%) solicitudes pendientes y 469 (33%) solicitudes abandonadas. Al 30 de junio de 2022, 571 (77%) de las patentes emitidas estaban activas y 171 (23%) habían expirado.
Tabla. Diez medicamentos de marca más vendidos en EE. UU. en 2021 sujetos a revisión de patentes.
| Tipo de medicamento | Uso clínico principal | fecha de aprobación de la FDA | Años de datos posteriores a la aprobación |
| Fármacos de molécula pequeña | |||
| Biktarvy (bictegravir/emtricitabina/tenofovir alafenamida) | VIH | 02/07/18 | 4.4 |
| Eliquis (apixabán) | Accidente cerebrovascular o embolia | 28/12/12 | 9.5 |
| Imbruvica (ibrutinib) | Cáncer | 13/11/13 | 8.6 |
| Revlimid (lenalidomida) | Mieloma múltiple | 27/12/05 | 16.5 |
| Productos biológicos | |||
| Enbrel (etanercept) | Artritis | 11/02/98 | 23.7 |
| Eylea (aflibercept) | Degeneración macular | 18/11/11 | 10.6 |
| Humira (adalimumab) | Artritis | 31/12/02 | 19.5 |
| Keytruda (pembrolizumab) | Cáncer | 09/04/14 | 7.8 |
| Stelara (ustekinumab) | Soriasis | 25/09/09 | 12.8 |
| Trulicity (dulaglutida) | Diabetes | 18/09/14 | 7.8 |
En el caso de los fármacos de molécula pequeña, la muestra contenía 279 patentes emitidas, de las cuales 102 (37%) figuraban en el Libro Naranja entre 1985 y 2022. En cuanto a los productos biológicos, la muestra contenía 463 patentes emitidas, de las cuales 88 (19%) figuraban en la versión del Libro Púrpura de junio de 2022. De todas las patentes emitidas, 242 (33%) han sido objeto de litigio desde el año 2000. El conjunto de datos incluía 12 patentes de fármacos de molécula pequeña que fueron objeto de litigio pero no figuraban en el Libro Naranja, y 56 patentes de productos biológicos que fueron objeto de litigio pero no figuraban en el Libro Púrpura.
Presentación de solicitudes de patente antes y después de la aprobación
Casi tres cuartas partes (1028 [72%]) de las 1429 patentes y solicitudes de patente se presentaron después de la aprobación de la FDA. Para los fármacos de molécula pequeña, 299 (58%) de las 517 solicitudes se presentaron después de la aprobación de la FDA. Para los productos biológicos, 729 (80%) de las 912 solicitudes se presentaron después de la aprobación de la FDA. Para 8 fármacos, la mayoría de las solicitudes de patente se presentaron después de la aprobación ( Figura 1 ). Los fabricantes presentaron una mediana (RIC) de 37 (10-61) solicitudes antes de la aprobación de la FDA y 99 (51-134) solicitudes después de la aprobación.
Figura 1. Patentes y solicitudes presentadas antes y después de la aprobación por la Administración de Alimentos y Medicamentos de los Estados Unidos (FDA) para 4 fármacos de molécula pequeña y 6 productos biológicos.
(Se abre en una pestaña nueva)
Para los fármacos de molécula pequeña, el número de solicitudes posteriores a la aprobación presentadas por año se mantuvo alto durante 5 años después de la aprobación de la FDA, luego disminuyó gradualmente ( Figura 2 A). La fecha mediana (RIC) de presentación de solicitudes para fármacos de molécula pequeña fue 1,2 años después de la aprobación de la FDA (1,9 años antes de la aprobación a 5,0 años después de la aprobación). Para los productos biológicos, el número de presentaciones posteriores a la aprobación de patentes y solicitudes alcanzó su punto máximo en el año 12 después de la aprobación ( Figura 2 B). La fecha mediana (RIC) de presentación de solicitudes para productos biológicos fue 5,9 años después de la aprobación de la FDA (0,8 años antes de la aprobación a 11,9 años después de la aprobación). Las prácticas de presentación variaron según el fármaco ( Figura 1 en el Suplemento 1 ).
Figura 2. Número promedio de patentes y solicitudes presentadas por año para 4 fármacos de molécula pequeña y 6 productos biológicos, y su estado al 30 de junio de 2022.
La línea vertical representa la fecha de aprobación de la Administración de Alimentos y Medicamentos de los Estados Unidos (FDA).
De las 742 patentes emitidas, 466 (63%) se presentaron después de la aprobación de la FDA: 48% (133 de 279) para fármacos de molécula pequeña y 72% (333 de 463) para productos biológicos. A los fabricantes se les concedió una mediana (RIC) de 28 (15-39) patentes presentadas antes de la aprobación de la FDA y 39 (15-58) patentes presentadas después de la aprobación, o una mediana general (RIC) de 77 (40-95) patentes totales por fármaco. Las carteras de patentes posteriores a la aprobación añadieron una mediana (RIC) de 7,9 (3,2-11,1) años de protección de patentes después de la expiración de las patentes previas a la aprobación.
Densidad de matorrales patentados
En el momento de la aprobación de la FDA, los medicamentos estaban protegidos por una mediana (RIC) de 16 (8-22) patentes activas. La densidad de patentes alcanzó su punto máximo 13 años después de la aprobación de la FDA. En ese momento, los medicamentos estaban protegidos por una mediana (RIC) de 42 (18-83) patentes activas, de las cuales el 66% (317 de 483) se presentaron después de la aprobación de la FDA. La protección de las patentes presentadas antes de la aprobación de la FDA alcanzó su punto máximo 5 años después de la aprobación de la FDA, cuando los medicamentos estaban protegidos por una mediana (RIC) de 23 (14-32) patentes presentadas antes de la aprobación de la FDA. La protección de las patentes presentadas después de la aprobación de la FDA alcanzó su punto máximo 15 años después de la aprobación, cuando los medicamentos estaban protegidos por una mediana (RIC) de 21 (11-40) patentes presentadas después de la aprobación. Para entonces, las patentes posteriores a la aprobación comprendían el 79% (347 de 438) de las patentes activas.
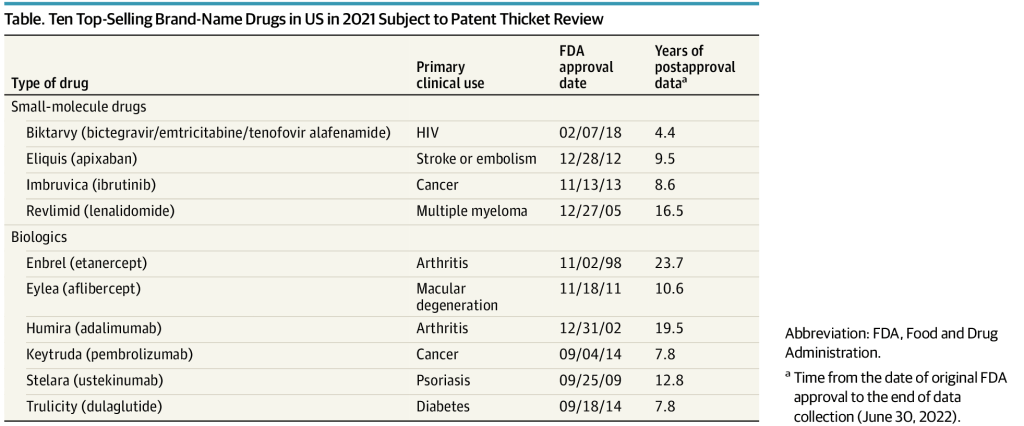
Los fármacos de molécula pequeña estaban protegidos por una mediana (RIC) de 17 (16-19) patentes en el momento de la aprobación de la FDA. La mediana (RIC) de patentes de fármacos de molécula pequeña alcanzó un máximo de 62 (22-96) patentes activas a los 12 años de la aprobación de la FDA, de las cuales el 52 % (118 de 226) se presentaron después de la aprobación. Posteriormente, la cantidad de patentes para fármacos de molécula pequeña se redujo rápidamente ( Figura 3 A).
Figura 3. Densidad promedio de matorrales de patentes para 4 fármacos de molécula pequeña y 6 productos biológicos.
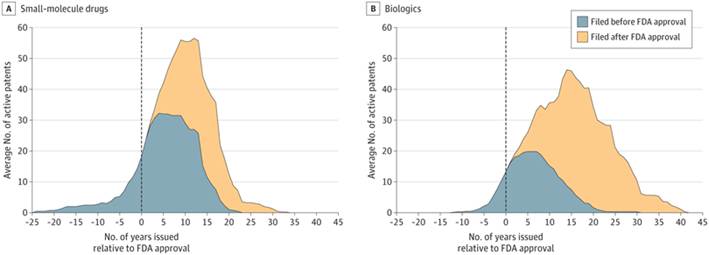
La línea vertical representa la fecha de aprobación de la Administración de Alimentos y Medicamentos de los Estados Unidos (FDA).
Por el contrario, los productos biológicos estaban protegidos por una mediana (RIC) de 8 (7-20) patentes en el momento de la aprobación de la FDA, y la mediana (RIC) de la red de patentes de productos biológicos alcanzó una densidad máxima de 41 (18-58) patentes activas a los 13 años de la aprobación, de las cuales el 76 % (197 de 260) provenían de patentes posteriores a la aprobación. Debido a que las solicitudes de patentes se distribuyeron a lo largo del tiempo, la red de patentes para productos biológicos se mantuvo densa durante más de 25 años ( Figura 3 B).
La composición de los conjuntos de patentes varió ampliamente para cada medicamento (Figura electrónica 2 en el Suplemento 1 ). Por ejemplo, el conjunto de patentes de Humira se expandió rápidamente aproximadamente 10 años después de la aprobación de la FDA, alcanzando 141 patentes activas. Este medicamento recibió su primera aprobación de la FDA en 2002, pero al 30 de junio de 2022, estaba protegido por 134 patentes activas, todas menos una de las cuales se presentaron después de la aprobación de la FDA. De manera similar, Enbrel tiene protección de patente hasta 2039, 41 años después de su aprobación de la FDA en 1998. El fabricante de este medicamento comenzó a presentar solicitudes de patentes de dispositivos 13 años después de la aprobación de la FDA. Al 30 de junio de 2022, casi la mitad (22) de las 45 patentes activas de Enbrel contenían reivindicaciones de dispositivos.
Los 10 fármacos que analizamos cuentan con algún tipo de protección de patente hasta una mediana (RIC) de 27,1 (22,9-32,6) años después de la aprobación de la FDA. Todas tienen patentes vigentes durante al menos otros 11 años y cuentan con otras solicitudes de patente pendientes.
Características de las patentes concedidas
De las 279 patentes emitidas para fármacos de molécula pequeña, 92 (33%) tenían reivindicaciones de composición química, 166 (60%) tenían reivindicaciones de método de uso, 22 (8%) tenían reivindicaciones de proceso o síntesis, 103 (37%) tenían reivindicaciones de formulación y 1 (0,4%) tenía una reivindicación de dispositivo. Sesenta y tres (43%) patentes previas a la aprobación y 19 (14%) patentes posteriores a la aprobación tenían más de un tipo de reivindicación.
De las 463 patentes emitidas para productos biológicos, 118 (26%) tenían reivindicaciones de composición química, 141 (31%) tenían reivindicaciones de método de uso, 147 (32%) tenían reivindicaciones de proceso o síntesis, 114 (25%) tenían reivindicaciones de formulación y 45 (10%) tenían reivindicaciones de dispositivo. Treinta y cuatro (26%) patentes previas a la aprobación y 52 (16%) patentes posteriores a la aprobación tenían más de un tipo de reivindicación.
Para fármacos de molécula pequeña y productos biológicos, la mayoría de las patentes con reivindicaciones de composición química se presentaron antes de la aprobación de la FDA. Las solicitudes de patentes con reivindicaciones de método de uso, proceso o síntesis, o formulación disminuyeron después de la aprobación de la FDA para fármacos de molécula pequeña, pero aumentaron después de la aprobación de la FDA para productos biológicos. Todas las patentes con reivindicaciones de dispositivos se presentaron después de la aprobación de la FDA ( Figura 4 ). De las 465 patentes posteriores a la aprobación, 86 (19%) tenían reivindicaciones de composición química, 189 (41%) tenían reivindicaciones de método de uso, 103 (22%) tenían reivindicaciones de proceso o síntesis, 127 (27%) tenían reivindicaciones de formulación y 46 (10%) tenían reivindicaciones de dispositivos.
Figura 4. Tipos de reivindicaciones en patentes concedidas para 4 fármacos de molécula pequeña y 6 productos biológicos.
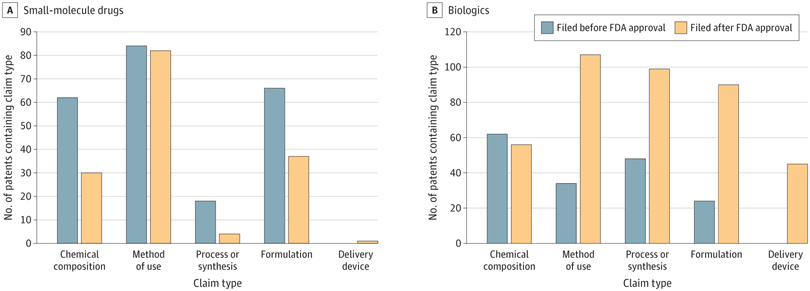
FDA significa Administración de Alimentos y Medicamentos de los Estados Unidos.
Discusión
Este estudio reveló que, entre los 10 medicamentos de marca más vendidos en EE. UU. en 2021, casi tres cuartas partes de las patentes y solicitudes de patente relacionadas con estos medicamentos se presentaron después de la aprobación de la FDA. Estos medicamentos estaban protegidos por una media de 77 patentes concedidas. En su punto álgido, estaban protegidos por un conjunto de patentes que comprendían una media de 42 patentes activas, de las cuales aproximadamente dos tercios se presentaron después de la aprobación de la FDA. La mayoría de las patentes posteriores a la aprobación cubrían aspectos de los medicamentos periféricos a sus principios activos, como los métodos de uso, las formulaciones y los procesos de fabricación. El conjunto de patentes solicitadas inicialmente después de la aprobación de la FDA plantea la cuestión de si estas patentes cubren descubrimientos verdaderamente novedosos e innovadores o si fueron diseñadas principalmente para bloquear la competencia oportuna de genéricos o biosimilares.
La proporción de solicitudes de patentes posteriores a la aprobación fue mayor para los productos biológicos (80 %) que para los fármacos de molécula pequeña (58 %). Las solicitudes de patentes para productos biológicos se presentaron más tarde, con una fecha media de presentación de 5,9 años después de la aprobación de la FDA, lo que supone más de 4 años después de la fecha media de presentación para los fármacos de molécula pequeña (1,2 años después de la aprobación de la FDA). El momento de la presentación de las solicitudes de patentes posteriores a la aprobación puede estar relacionado estratégicamente con la expiración prevista de las exclusividades de mercado no basadas en patentes, que difieren para los fármacos de molécula pequeña y los productos biológicos. Estas exclusividades de mercado están previstas en leyes distintas de la Ley de Patentes que impiden a la FDA aprobar productos de la competencia incluso en ausencia de una patente. Por ejemplo, en virtud de la Ley de Competencia de Precios de Medicamentos y Restauración del Plazo de Patentes de 1984, los nuevos fármacos de molécula pequeña pueden recibir al menos 5 años de exclusividad antes de que se puedan presentar solicitudes de genéricos. 16 En nuestro estudio, encontramos que la presentación de solicitudes de patentes posteriores a la aprobación de medicamentos de moléculas pequeñas alcanzó su punto máximo aproximadamente en los primeros 5 años después de la aprobación de la FDA, ya que los fabricantes de medicamentos estaban creando sus complejos entramados de patentes durante los años de exclusividad regulatoria. Por el contrario, los productos biológicos reciben 12 años de exclusividad regulatoria de mercado antes de que un biosimilar pueda ser considerado para la aprobación de la FDA bajo la Ley de Competencia e Innovación de Precios de Productos Biológicos de 2009 (BPCIA). 17 Entre los productos biológicos de nuestra muestra, observamos un pico en la presentación de solicitudes de patentes en el año 12 después de la aprobación de la FDA.
La mayoría de las patentes presentadas después de la aprobación de la FDA entre los 10 medicamentos de nuestra muestra cubrían métodos de uso, formulaciones y métodos de fabricación, en lugar de variaciones en los ingredientes activos (como derivados). Otras investigaciones han demostrado que las patentes sobre métodos de uso u otros aspectos periféricos contribuyen a la mayoría de los litigios presentados contra los fabricantes de biosimilares que buscan la autorización de comercialización. 18 Además, investigaciones previas han demostrado que las patentes que protegen las formulaciones y los métodos de uso suelen ser patentes más débiles y menos propensas a mantenerse después de ser impugnadas en litigios por fabricantes de medicamentos genéricos. 19 En nuestro estudio, los productos biológicos tenían una mayor proporción de reivindicaciones de proceso o síntesis que los medicamentos de molécula pequeña, particularmente cuando las patentes se presentaron después de la aprobación de la FDA. Esto podría explicarse por los procesos de fabricación más complejos de los productos biológicos que brindan la oportunidad de presentar más patentes con respecto a los métodos de fabricación, que a menudo no son específicos del producto. 20 Nuestros resultados también reflejan la creciente práctica de patentar dispositivos de administración de productos biológicos. 21 Si bien las patentes de dispositivos médicos representaron solo el 10% de las patentes de productos biológicos en nuestra muestra, todas se presentaron después de la aprobación de la FDA.
La densidad de la maraña de patentes alcanzó su punto máximo aproximadamente 13 años después de la aprobación de la FDA. En ese momento, los medicamentos estaban protegidos por una mediana de 42 patentes activas; los fabricantes de medicamentos genéricos o biosimilares de la competencia debían evaluar cada patente para determinar si su producto infringía la protección de patente. Independientemente de la validez de cada patente, la densidad general de la maraña de patentes puede disuadir la competencia 22 , 23 al aumentar el costo percibido de entrada al mercado. Los competidores deberán predecir la probabilidad de que cada patente se considere válida, ejecutable e infringida, 24 y sopesar los costos de impugnarla administrativamente o mediante litigios. 23 Este proceso podría ser aún más complejo para los fabricantes de biosimilares, quienes podrían tener que identificar e impugnar patentes de fabricación no específicas del producto, ejercidas por fabricantes de marcas reconocidas. 20 Si bien la maraña de patentes en la industria farmacéutica se utiliza estratégicamente para prevenir la competencia (por ejemplo, en el caso de adalimumab [Humira]), 25 impugnarla es una tarea ardua. 26
Investigaciones previas han revelado perfiles de densidad de cúmulos de patentes similares para 4 de los fármacos incluidos en esta muestra. 27 Sin embargo, el número de patentes activas en el pico de densidad de cúmulos en nuestro estudio es mayor que el reportado anteriormente. Descubrimos que los cúmulos de patentes para productos biológicos contenían más patentes en general y se mantuvieron densos durante más tiempo que los de fármacos de molécula pequeña. Estos resultados concuerdan con datos previos que indican que los cúmulos de patentes son más grandes y la mediana de exclusividad de mercado es más larga para los productos biológicos que para los fármacos de molécula pequeña (21,5 frente a 14,4 años). 28
Limitaciones
Nuestro estudio tiene varias limitaciones. Analizamos el panorama de patentes de 10 medicamentos superventas, y estos resultados podrían no ser generalizables a medicamentos para enfermedades raras o productos de baja facturación. Los datos posteriores a la aprobación abarcaron un período más largo para algunos medicamentos que para otros. Los fabricantes han seguido presentando solicitudes de patentes después de que dejamos de recopilar datos, y algunas solicitudes actualmente pendientes podrían convertirse en patentes concedidas. Por ello, podríamos subestimar el impacto de las patentes posteriores a la aprobación. Dado que el sitio web de la USPTO podría estar incompleto, es posible que tampoco hayamos tenido en cuenta todas las extensiones de la vigencia de las patentes, lo que podría subestimar el impacto de las patentes posteriores a la aprobación. Además, es posible que nuestra lista de patentes fuera demasiado extensa, ya que incluía más patentes que las que figuran en el Libro Naranja o el Libro Púrpura. Ninguno de los compendios publicados por la FDA es exhaustivo ni contiene todas las patentes relacionadas con un producto; en particular, el Libro Púrpura solo ha comenzado a incluir patentes recientemente y se limita a aquellas presentadas en ciertas etapas de litigios específicos de fabricantes relacionados con la BPCIA, mientras que el Libro Naranja excluye las patentes de métodos de fabricación. Existen diversas técnicas que los terceros pueden utilizar para evitar llegar al punto en que sus listas de patentes queden sujetas al requisito de presentarlas al Libro Púrpura de la FDA. 29 Del mismo modo, si el fabricante de biosimilares decide operar fuera del contexto de la BPCIA y lanzar el producto asumiendo el riesgo, no existe ningún requisito para que el fabricante de productos biológicos de marca publique sus patentes en el Libro Púrpura.
Finalmente, no revisamos de forma independiente cada patente o solicitud de patente para determinar su relevancia o la solidez de sus reivindicaciones. Sin embargo, sí identificamos patentes en el conjunto de datos que fueron objeto de litigio pero que no figuraban en el Libro Naranja ni en el Libro Púrpura, lo que indica que las patentes relevantes van más allá de las divulgadas a la FDA. En nuestro análisis de las prácticas de presentación, incluimos tanto las patentes concedidas como las solicitudes de patente. El análisis de las solicitudes de patente puede ser poco fiable, ya que las reivindicaciones pueden cambiar con el tiempo y, en última instancia, no estar relacionadas con el medicamento. No obstante, las solicitudes de patente seguirán siendo relevantes para los posibles competidores que busquen entrar en el mercado, ya que tendrán que predecir si la solicitud de patente dará lugar finalmente a una patente concedida. 24 Además, algunas de las patentes pueden tener reivindicaciones limitadas o ser inválidas. Sin embargo, esto refleja el panorama de patentes que los fabricantes de medicamentos genéricos o biosimilares de la competencia tendrán que evaluar al considerar si entrar en el mercado. 30
Conclusión
En este estudio, entre los 10 medicamentos recetados más vendidos en EE. UU. en 2021, las patentes presentadas después de la aprobación de la FDA y que contenían reivindicaciones periféricas al principio activo contribuyeron a la formación de densas marañas de patentes, lo que dificulta considerablemente el acceso de los consumidores a opciones más asequibles. Estas complejas redes de propiedad intelectual complican no solo la introducción de genéricos y biosimilares en el mercado, sino también la innovación en la industria farmacéutica. Es necesario un examen minucioso de las solicitudes de patente y de las patentes presentadas después de la aprobación de la FDA para facilitar la competencia oportuna de genéricos y biosimilares, asegurando así que los pacientes puedan beneficiarse de tratamientos más económicos y efectivos. Una revisión crítica de estas prácticas podría promover un entorno más equitativo en el acceso a medicamentos esenciales y fomentar la sostenibilidad del sistema de salud en general.

{kind=link}
{kind=link}