Eckhardt, H., Felgner, S., Dreger, M. et al. Utilization of innovative medical technologies in German inpatient care: does evidence matter?. Health Res Policy Sys 21, 100 (2023). https://doi.org/10.1186/s12961-023-01047-w
El progreso tecnológico relacionado con la salud desempeña un papel importante en la mejora de los resultados sanitarios. Sin embargo, las nuevas tecnologías también pueden conllevar riesgos para los pacientes y usuarios [1, 2]. Desde el escándalo de Contergan (talidomida) en la década de 1950 [3], el acceso al mercado de los productos farmacéuticos está altamente regulado y, por lo general, requiere una evaluación clínica exhaustiva.
El proceso de aprobación de nuevos productos sanitarios en el contexto europeo está descentralizado e implica la verificación de la conformidad de un producto con el marco normativo de la Unión Europea (UE), principalmente en lo que respecta a su uso previsto y su seguridad.
En 2007 se introdujo la necesidad de realizar investigaciones clínicas para determinar la eficacia y la seguridad de los nuevos productos sanitarios implantables o de alto riesgo para su aprobación en el mercado (Directiva 2007/47/CE [4]). A raíz de los reiterados informes de daños a los pacientes, el Reglamento sobre productos sanitarios (MDR) [Reglamento (UE) 2017/745 [5]), cuyo objetivo era reformar el marco regulador de la UE sobre productos sanitarios (Directiva 93/42/CEE [6] y Directiva 90/385/CEE [7]), introdujo por primera vez el término «beneficio clínico» como criterio para la aprobación de productos sanitarios [8]]. Por el contrario, el proceso de aprobación previa a la comercialización por parte de la Administración de Alimentos y Medicamentos (FDA) en los Estados Unidos (EE. UU.) ha requerido evidencia de ensayos clínicos para determinar la efectividad y seguridad de los dispositivos médicos innovadores de alto riesgo desde principios de la década de 1990 [9,10,11].
Una vez que han sido aprobadas para su comercialización, el camino hacia el reembolso de las tecnologías médicas en los sistemas sanitarios europeos suele variar [12,13,14]. Si bien la evaluación de tecnologías sanitarias (ETS) para determinar la (coste)eficacia comparativa de las nuevas tecnologías suele ser un requisito previo para los productos farmacéuticos en el sector ambulatorio, no siempre es así para otras tecnologías o para el entorno hospitalario [13]. Sin embargo, los productos sanitarios innovadores, de alto riesgo y alto coste son objeto con mayor frecuencia de ETS vinculadas al reembolso [15].
Este no es siempre el caso en Alemania, donde tradicionalmente todas las nuevas tecnologías para el diagnóstico o el tratamiento (Neue Untersuchungs- und Behandlungsmethode, NUB) han sido reembolsables en la atención hospitalaria, a menos que el Comité Mixto Federal (FJC; § 137c Libro V del Código Social Alemán [SGB V]). Cuando los costes de estos NUB aún no pueden ser suficientemente contabilizados por un grupo relacionado con el diagnóstico (GRD) existente, los hospitales pueden solicitar al Instituto Alemán del Sistema de Remuneración Hospitalaria (InEK) el permiso para negociar pagos extrapresupuestarios de un año para cada hospital (también denominados «pagos por innovación») con las aseguradoras de salud [16, 17]. Una vez que estas nuevas tecnologías han sido incluidas en la Clasificación de Procedimientos Alemana [16], su financiación adecuada basada en los costes se consigue a través de pagos suplementarios (fijos o negociables) considerados en las negociaciones presupuestarias anuales entre los hospitales y las aseguradoras de salud, la división de un GRD existente o la creación de uno nuevo. Los pagos suplementarios fijos conllevan un precio nacional uniforme por caja publicado en el apéndice del catálogo DRG; Cuando no se puede determinar un precio nacional uniforme, los hospitales pueden obtener pagos suplementarios negociables individuales (confidenciales) con un importe mínimo de 600 euros por caso [17].
En el contexto de la falta de requisitos de HTA antes del reembolso y de los incentivos financieros descritos, es importante para la calidad de la atención y la relación calidad-precio indagar si la implementación de tecnologías innovadoras en la atención hospitalaria alemana se basa en la evidencia clínica. Las investigaciones previas sobre la influencia de la evidencia o los mecanismos de financiación en la implementación y difusión se han centrado en gran medida en tecnologías individuales [17,18,19] o en decisiones de cobertura [20].
El objetivo general de este trabajo era doble: en primer lugar, investigar el tipo de evidencia que estaba disponible en el momento de la introducción de tecnologías innovadoras seleccionadas en la atención hospitalaria alemana y cómo evolucionó esta evidencia a lo largo del tiempo. En segundo lugar, se buscó explorar el vínculo entre la evidencia científica disponible, como ensayos clínicos, revisiones sistemáticas o informes de evaluación de tecnologías sanitarias, y la difusión de estas tecnologías.
Este análisis exploratorio constó de tres etapas: (i) la identificación sistemática de la evidencia científica con respecto a la seguridad y eficacia/efectividad de cada tecnología, (ii) la descripción del conjunto de evidencia disponible para cada tecnología en el momento de su introducción y su desarrollo a lo largo del tiempo; y (iii) la investigación de la relación estadística entre la evidencia identificada y la utilización de tecnologías por parte de los hospitales. Además, se investigó el impacto potencial de las guías clínicas, los mecanismos de financiación y los avisos de seguridad (advertencias y retiros) en la utilización de estas tecnologías en función del paso (ii) anterior, para facilitar una interpretación más contextualizada.
Métodos
Tecnologías seleccionadas y datos sobre la utilización
El conjunto de tecnologías admisibles comprendía aquellas para las que se concedieron entre 2005 y 2012 permisos solicitados para pagos extrapresupuestarios a personas hospitalarias. Esta ventana de tiempo garantizó la disponibilidad de datos durante al menos cinco años antes del inicio de esta investigación en 2018 (es decir, el período de observación de este estudio: 2005-2017). Las curvas de utilización de cada tecnología se trazaron sobre la base de los datos identificados a través de los respectivos códigos de procedimiento del conjunto de datos estadísticos de DRG. Este conjunto de datos es puesto a disposición por el Centro de Datos de Investigación (RDC) de la Oficina Federal de Estadística y las Oficinas de Estadística de los Estados Federados [21] previa solicitud formal y acuerdo previo. A los efectos de este análisis, el financiador proporcionó datos sobre el número de procedimientos de hospitalización por tecnología, por hospital y por año. Se incluyó una tecnología en el análisis si cumplía los siguientes criterios: entre 2005 y 2012, al menos diez hospitales solicitaron y obtuvieron autorización para recibir pagos extrapresupuestarios entre hospitales y personas durante un mínimo de un año, el número de procedimientos y el número de hospitales que utilizaban la tecnología estaban disponibles durante al menos cuatro años, y se habían realizado al menos 100 procedimientos durante un mínimo de un año. Sobre la base de estos criterios, se preseleccionaron 59 tecnologías y se aplicó un análisis cuantitativo de conglomerados para reducir los diferentes tipos de perfiles de utilización. Finalmente, 25 tecnologías fluyeron en el análisis (este proceso se describe en detalle en otra parte [22]). Este enfoque tenía como objetivo garantizar que la muestra final incluyera tanto tecnologías con grandes volúmenes de utilización como aquellas con inconsistencias en los patrones de utilización. Los códigos de procedimiento para las 25 tecnologías y sus descripciones figuran en el apéndice 1 del fichero adicional 1.
Búsqueda de información sobre financiamiento, retiros del mercado y advertencias de seguridad
Las fuentes de información sobre la financiación, las retiradas y las advertencias de seguridad por tecnología se enumeran en el Apéndice 2 del Archivo Adicional 1.
Búsqueda de evidencia clínica
En 2019 se realizó una búsqueda bibliográfica sistemática en PubMed, Medline (vía OVID), Embase (vía OVID) y la Biblioteca Cochrane. El desarrollo de estrategias de búsqueda para cada tecnología incluida se basó en el marco PICO (población, intervención, comparación, resultados). Para definir los términos de búsqueda, se consultaron las evaluaciones del Servicio Médico de la Asociación Nacional de Cajas de Seguro de Salud y la Clasificación de Procedimientos, centrándose en la intervención (términos específicos de la tecnología, nombres de productos y fabricantes) y la indicación (las estrategias de búsqueda específicas de la tecnología están disponibles a pedido). Se realizaron búsquedas suplementarias en las listas de referencias de las revisiones sistemáticas incluidas, los registros de ensayos clínicos, las bases de datos de ETS y las bases de datos de guías clínicas (detalles en el Archivo adicional 1, Apéndice 2). Los archivos EndNote X9 se crearon por tecnología y se utilizaron para la eliminación de duplicados [23], después de importar los resultados de la búsqueda y para la documentación del proceso de selección.
Selección de la evidencia
Se elaboraron criterios generales de inclusión y exclusión basados en el marco PICO para seleccionar las pruebas pertinentes para cada tecnología (todos los detalles figuran en el Archivo adicional 1, Apéndice 3, cuadro S3.1). Los estudios publicados y no publicados pertenecientes a los niveles de evidencia (LoE) 1-4 según la definición del FJC (2. Se incluyó el Capítulo §11 [3], reglas de procedimiento de FJC) [24], mientras que los estudios pertenecientes a LoE 5 fueron documentados, pero no analizados más a fondo:
- 1a Revisiones sistemáticas de ensayos controlados aleatorizados
- 1b Ensayos controlados aleatorizados (ECA)
- 2a Revisiones sistemáticas de ensayos controlados no aleatorizados
- 2b Ensayos controlados prospectivos no aleatorizados (N-ECA)
- 3 Ensayos controlados retrospectivos
- 4 Series de casos y otros ensayos de un solo grupo
- 5 Informes de casos, etc.
Las publicaciones que comenzaron dos años antes de la primera documentación de los procedimientos hospitalarios (más temprano en 2003) y hasta 2017 fueron elegibles para su inclusión.
La evidencia se seleccionó de acuerdo con los métodos de revisión rápida de la Colaboración Cochrane [25]. Después de la eliminación de duplicados, se extrajo una muestra aleatoria del 10% de las citas (mín. 100) por tecnología a través de RStudio (versión 1.4.1717). Dos investigadores examinaron de forma independiente la muestra y seleccionaron las citas relevantes. En caso de discrepancias, se discutieron y ajustaron los criterios de inclusión y exclusión, involucrando a un tercer investigador si fuera necesario. Las citas restantes fueron examinadas por una persona en función de los criterios ajustados. Cada paso de la revisión se documentó según lo recomendado por la declaración PRISMA [26].
La selección de la evidencia identificada en los registros de ensayos, las guías y las bases de datos de HTA, y el proceso de extracción de datos y la evaluación del riesgo de sesgo (RoB) se describen en los apéndices 4, 5 y 6 del archivo adicional 1, respectivamente.
Categorización de los resultados del estudio
Para cada publicación, las conclusiones de los autores se extrajeron del resumen y del texto principal y se clasificaron en «positivas», «negativas», «neutrales» o «no concluyentes» de acuerdo con el esquema de categorización descrito en la Fig. 1. En caso de que no se dispusiera de una sección de conclusiones en el texto principal, la categorización se realizó con base en el resumen de resultados de la sección de discusión y del resumen.
Síntesis y análisis estadístico
Para explorar (a) las características del cuerpo de evidencia disponible para cada una de las tecnologías incluidas en el momento de su introducción y a lo largo del tiempo y (b) la posible relación entre la evidencia y los patrones de utilización, se realizó un análisis descriptivo y estadístico.
Análisis descriptivo: El cuerpo de evidencia identificado fue analizado descriptivamente. Cada publicación se consideró como un punto de datos independiente. Las recomendaciones de las guías clínicas, la información sobre la financiación y los avisos de seguridad se sintetizaron narrativamente para cada tecnología y se discuten en combinación con otros hallazgos.
Evaluación empírica de la relación entre la utilización y los resultados de la evidencia: Para evaluar si la utilización de una tecnología a lo largo del tiempo sigue la evidencia clínica disponible, se agregó una nueva variable X, «resultados del cuerpo de evidencia disponible» en el año t y la tecnología j. La variable incorpora todos los análisis comparativos identificados (LdE 1-3) de una tecnología publicada hasta el año t inclusive, ponderados por categoría de resultados del estudio (positivos, negativos, neutros) y SdE (LdE 1-3). La variable representa los resultados predominantes del conjunto de pruebas disponibles a partir de dos años antes del inicio de la utilización en Alemania, acumuladas a lo largo de todos los años de utilización hasta el año t. Los datos de la variable de resultado «utilización» están representados por el número de procedimientos hospitalarios en el año t y la tecnología j.
Debido a la estructura de datos agrupados y para tener en cuenta los efectos específicos de la tecnología, se aplicó un modelo de efectos mixtos (modelo multinivel) para dos niveles de datos [29, 30]. Se describe con más detalle en el Archivo adicional 1, Apéndice 7. El objetivo de la función de regresión es estimar si el desarrollo de la utilización sigue la dirección de los resultados del estudio, pero no explicar toda la varianza; Esto no sería posible si se incluyera solo una variable explicativa, pero la mayoría de los otros factores potencialmente influyentes [31] son difíciles de cuantificar. El análisis de regresión se realizó utilizando Rstudio (versión 1.4.1717) y el paquete lme4.
Resultados
Resultados de la búsqueda de evidencias
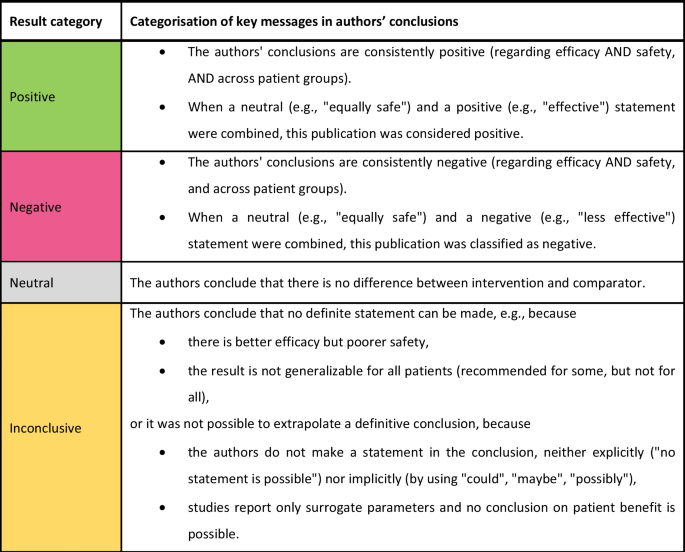
Todas las búsquedas arrojaron números de citas en el rango de cuatro dígitos. El número de publicaciones incluidas en el análisis final (LoE 1-4) oscila entre dos (catéter balón farmacológico en vasos abdominales, DCB-AV) y 498 (implante de válvula aórtica transcatéter, TAVI) (Fig. 2). Archivo adicional 1, Apéndice 8, tabla 8.1 muestra los resultados de las búsquedas y selección por tecnología, base de datos bibliográfica y etapa dentro del proceso de selección.
Los resultados de las búsquedas de literatura gris (guías clínicas, informes de ETS, entradas en el registro de ensayos y notificaciones de seguridad) se presentan en el Archivo adicional 1, Apéndice 8, tabla 8.2. A través de tecnologías y años, se identificaron 40 informes de HTA y 40 guías clínicas. El número de informes de HTA por tecnología varía entre cero y 12. Se identificaron recomendaciones de guías clínicas para 19 tecnologías; el número de guías (y sus actualizaciones) varía entre cero y 13 (resección transuretral asistida por fluorescencia, guía clínica F-TUR con 12 actualizaciones anuales entre 2006 y 2017). Otros resultados sobre las guías clínicas se presentan en el Archivo Adicional 1, Apéndice 12.
Se identificó al menos una notificación de seguridad para 12 tecnologías en Alemania y para otras dos tecnologías a nivel internacional. El número de notificaciones de seguridad en Alemania oscila entre una (asistencia pulmonar extracorpórea sin bomba/asistencia pulmonar intervencionista, PECLA/iLA) y 12 (TAVI). A nivel internacional, el TAVI y el andamio vascular biorreabsorbible en vasos coronarios (BVS) son las tecnologías con más notificaciones (74 notificaciones para TAVI y 48 para BVS). Se identificó al menos un retiro del mercado para siete tecnologías en Alemania y para seis tecnologías adicionales a nivel internacional.
Características del acervo probatorio
La composición del conjunto de pruebas por tecnología se muestra en la Fig. 2A. Para casi todas las tecnologías, la mayor parte de la evidencia consiste en series de casos y otros estudios no comparativos; estos diseños constituyen más de la mitad de todas las publicaciones identificadas en todas las tecnologías (943/1840) (Fig. número arábigoB y C). Sólo 213/1840 (12%) de las publicaciones incluidas informan resultados de 130 ECA individuales. No se identificó ningún ECA para seis de las 25 tecnologías. Para las 19 tecnologías restantes, el número de ECA con al menos una publicación por tecnología varía de una (p. ej., PECLA/iLA) a 19 (catéter balón recubierto de fármaco en vasos coronarios; DCB-CV). La mayoría de los ECA identificados muestran un RoB alto. Solo para cinco tecnologías, se identificó al menos un ECA con RoB bajo (Archivo adicional 1, Apéndice 9, Fig. S9.1).
El número de revisiones sistemáticas e informes de HTA oscila entre cero (p. ej., desviador de flujo en los vasos de la parte superior de la pierna, FD-ULV) y 37 (TAVI). El número de revisiones sistemáticas es mayor que el número de ECA para la mayoría de las tecnologías. Para algunas tecnologías, hubo varias revisiones sistemáticas que analizaron el mismo grupo de ECA (p. ej., trombectomía mecánica endovascular intracraneal, MT). Se pueden encontrar más detalles sobre las características del estudio en el Archivo adicional 1, Apéndice 9.
Evolución del acervo probatorio a lo largo del tiempo
En los primeros años de utilización documentada, el número de publicaciones disponibles es bajo para casi todas las tecnologías de la muestra. Los resultados de los diseños de estudios no comparativos (LoE 4) suelen dominar el panorama. A pesar del aumento de la proporción de pruebas de la LoE 1-3 a lo largo del período de observación (Archivo adicional 1, Apéndice 10, Fig. S10.1), el número de tales publicaciones sigue siendo inferior al del Nivel 4 para la mayoría de las tecnologías (Archivo adicional 1, Apéndice 10, Fig. S10.2). Se puede observar un desfase sustancial (hasta 9 años) entre el primer año de utilización y la publicación de los primeros resultados de los ECA para varias tecnologías (Archivo adicional 1, Apéndice 9, Fig. S9.1).
Resultados de la evidencia sobre tecnologías innovadoras
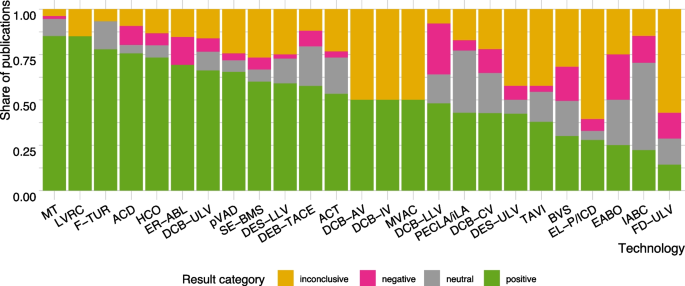
La Figura 3 muestra la proporción de publicaciones con resultados positivos, negativos, neutrales y no concluyentes por tecnología. En el caso de algunas tecnologías, la proporción de publicaciones no concluyentes es relativamente alta; en particular, las series de casos y otros estudios de un solo grupo no siempre cayeron en la categoría positiva, negativa o neutra debido a la ambigüedad de las conclusiones (p. ej., mejor eficacia pero menor seguridad) (Archivo adicional 1, Apéndice 11, Fig. S11.1). En el Archivo Adicional 1, Apéndice 11, Figs. S11.2 y 11.3 (A-C). Los resultados negativos tendieron a observarse con mayor frecuencia entre los estudios con un grupo de comparación. Por el contrario, los estudios de LoE 4 tendieron a concluir positivamente con más frecuencia.
Utilización de tecnologías innovadoras
La utilización observada de las tecnologías en la muestra de estudio, medida por el número facturado de procedimientos por año, se muestra en la Fig. 4. El número máximo de procedimientos por año oscila entre 138 (2014, stents metálicos autoexpandibles en vasos coronarios, SE-BMS) y 42.203 (2017, anticoagulación con citrato durante la diálisis, ACD). El número de años con procedimientos hospitalarios reembolsados identificados a través de códigos de procedimiento específicos en el período de observación varió de cuatro (SE-BMS) a 13 (Cardiac event recorder after ablative measures for atrial fibrillation/atrial tachycardia—ER-ABL), con una mediana de 10 (2008).

Financiación de tecnologías innovadoras
Como se muestra en la Fig. 4, el comienzo de la utilización está vinculado a la autorización para negociar pagos extrapresupuestarios con los fondos del seguro médico para la mayoría de las tecnologías. Las excepciones son ER-ABL, F-TUR y anuloplastia de la válvula mitral con pinza (MVAC), para las cuales la utilización comienza antes. En el caso de la mayoría de las tecnologías, el fin de la autorización para negociar pagos extrapresupuestarios coincide con el comienzo de la siguiente etapa de reembolso. Al final del período de observación, se aplicaban pagos suplementarios negociables o fijos para 12 de las 25 tecnologías, mientras que siete estaban incluidas en un DRG. El cambio de reembolso se produjo después de uno a siete años. Para las cinco tecnologías restantes (oclusión endoaórtica con balón con circulación extracorpórea, EABO, FD-ULV, espirales bioactivas para el tratamiento de aneurismas intracraneales, IABC, MVAC, SE-BMS), la financiación no cambió durante el período de observación.
Relación entre la utilización y otros factores
La regresión multinivel mostró una relación estadísticamente significativa entre la dirección de la evidencia y la dirección de utilización (Tabla 1). Los diferentes órdenes de magnitud en la utilización de las distintas tecnologías, como resultado principalmente de la prevalencia variable de las afecciones subyacentes, pueden explicar la gran varianza. La alta correlación intraclase (CCI) confirma la idoneidad del enfoque agrupado [30, 32].
Tabla 1 Relación entre el conjunto de pruebas disponibles y la utilización: resultados de la regresión multinivel
La Figura 4 ilustra la concordancia entre la utilización anual observada y la utilización prevista sobre la base de la evidencia (valores ajustados) por tecnología. Para la mayoría de las tecnologías, el aumento de la utilización va acompañado de pruebas positivas (p. ej., TAVI) o la disminución de la utilización va acompañada de pruebas negativas (p. ej., BVS). Por el contrario, los valores pronosticados no están en línea con los valores observados para siete tecnologías (p. ej., reducción del volumen pulmonar mediante la inserción de espirales, LVRC); Esto implica que otros factores, además de la evidencia, tuvieron una mayor influencia en la utilización.
Como se puede ver en la Fig. 4, no se observan pautas coherentes en cuanto a la relación entre las tendencias de utilización y los cambios en la financiación.
El impacto de las notificaciones de seguridad y los retiros del mercado es difícil de analizar, sobre todo porque hay varios productos disponibles para la mayoría de las tecnologías de la muestra. En el caso de las tecnologías con un máximo de dos productos identificados y un recuerdo identificado (extracción con láser excimer de electrodos de marcapasos y desfibrilador, EL-P/ICD [33] en 2012, diálisis con membrana de diálisis de alto corte, HCO [34] en 2011, PECLA/iLA [35] en 2006), no se observa ninguna relación. En el caso de BVS, las advertencias de seguridad que restringían el uso a ciertos recipientes [36] y a instalaciones seleccionadas que participaban en registros clínicos [37] iban acompañadas de evidencia negativa seguida de una disminución en la utilización.
Discusión
Este trabajo evaluó la relación entre la utilización de 25 tecnologías para diferentes sistemas anatómicos en la atención hospitalaria alemana y la evidencia clínica disponible, los cambios en la financiación y la información de seguridad.
El número de publicaciones incluidas por tecnología osciló entre dos y 498, y las series de casos y otros diseños no comparativos constituyeron la mayor parte del cuerpo de evidencia. Aunque esto refleja el interés de los médicos en compartir su experiencia con una determinada tecnología en un entorno del mundo real, estos estudios no proporcionan una base adecuada para concluir sobre el valor comparativo de una tecnología. Para siete de las 25 tecnologías, se identificó un máximo de una publicación de cualquiera de los dos niveles de evidencia más altos (revisiones sistemáticas de ECA o ECA individuales) a pesar de las altas cifras de utilización. De hecho, para la mayoría de las tecnologías incluidas hubo pocos ECA, predominantemente con RoB alto. Un gran volumen de publicaciones no necesariamente supedita la solidez del cuerpo de evidencia en el beneficio de una tecnología. Del mismo modo, la disponibilidad de múltiples revisiones sistemáticas de estudios comparativos (LoE 1a/2a) para la misma tecnología no da lugar necesariamente a una ganancia de información. Por ejemplo, en este trabajo se identificaron varias revisiones/metaanálisis sistemáticos sobre recuperadores de stents para la trombectomía mecánica en el accidente cerebrovascular agudo, publicado en un plazo de dos años, y que combina los mismos seis ECA. Es más, los hallazgos de las revisiones sistemáticas son tan sólidos como lo permitan los estudios incluidos en la revisión.
La tendencia hacia una mayor LoE a lo largo del tiempo no siempre fue observable. En el caso de varias tecnologías, no se materializó un conjunto sólido de pruebas (p. ej., al menos un ECA con bajo riesgo de sesgo) durante varios años o incluso hasta el final del período de observación. De una tecnología a otra, varió el número de años y el número de procedimientos realizados antes de que se dispusiera de pruebas científicas adecuadas. El tipo de aprobación de la FDA, que está vinculado a los requisitos de generación de evidencia posterior a la comercialización y, por lo tanto, podría haber influido en el número y tipo de estudios disponibles [38, 39], no se investigó más a fondo. Sin embargo, parece que la falta de una evaluación obligatoria de las prestaciones antes del reembolso puede exponer a los pacientes a un daño indebido (incluida la falta de prestaciones) y al sistema de salud a un gasto ineficiente. Otros países europeos, como Francia [40] y el Reino Unido [41], han establecido vías de evaluación comparativa de la eficacia de las nuevas tecnologías antes del reembolso. En 2016 se estableció una vía de evaluación para ciertas tecnologías innovadoras de alto riesgo en la atención hospitalaria alemana (§137h SGB V), que obliga a los hospitales que buscan negociar pagos extrapresupuestarios por primera vez a proporcionar al FJC la evidencia disponible sobre la efectividad. Esto no se aplicó a ninguna de las tecnologías de la muestra.
Podría demostrarse una relación entre la evidencia y la utilización para algunas tecnologías, pero no para todas. Hasta cierto punto, esto era de esperar, ya que hay muchos factores que influyen en la adopción de nuevas tecnologías en las organizaciones de atención médica que no podrían ser explicados en el modelo de regresión. El número disponible de tecnologías alternativas, la experiencia del operador, la facilidad de uso, la cultura organizacional, las creencias y preferencias individuales de los operadores, así como la demanda de los pacientes [42, 43] pueden desempeñar un papel en el proceso de difusión. También es pensable que los altos costos iniciales de adquisición conduzcan al uso continuo de la tecnología a pesar de la disponibilidad de mejores alternativas. Por último, incluso si una tecnología es reembolsable y tiene un perfil de evidencia positivo, es posible que las aseguradoras no estén dispuestas a aceptar pagos extrapresupuestarios o suplementarios que cubran todos los costos.
La relación entre la utilización y la financiación, así como la relación entre la utilización y las notificaciones de seguridad se exploraron de manera cualitativa, sin resultados claros. Sin embargo, esto no excluye totalmente la posibilidad de que tales relaciones existieran para cualquiera de las tecnologías. Este estudio no fue diseñado para predecir cómo podría haber sido la utilización si la financiación no hubiera cambiado con el tiempo, pasando de los tipos de financiación más inseguros (pagos extrapresupuestarios) a tipos de financiación más seguros (por ejemplo, una descripción adecuada en un GRD). Para al menos una tecnología, los avisos y restricciones de seguridad podrían haber fortalecido el efecto de la evidencia clínica, pero esta relación también resultó imposible de evaluar completamente con este diseño de estudio.
Para garantizar la calidad de la atención, es importante que las tecnologías reembolsadas sean seguras y eficaces. El primer paso para lograrlo es contar con procesos regulatorios que solo permitan que dichos dispositivos médicos ingresen al mercado. Los cambios introducidos por el MDR, que entraron en vigor en mayo de 2021 [5, 8], podrían reducir el número de años de utilización sin pruebas sólidas en la atención hospitalaria alemana y en el resto de Europa (y el desfase temporal entre la certificación CE y la aprobación de la FDA [44]), pero esto también dependerá de cómo se implemente la regulación.
El segundo paso consiste en evaluar la eficacia comparativa o incluso la rentabilidad de las nuevas tecnologías antes del reembolso. Todos los sistemas de salud deben equilibrar el acceso oportuno con la certeza sobre la seguridad y la eficacia de una innovación, al tiempo que distribuyen los recursos limitados de manera inteligente. En el caso de las nuevas tecnologías que se muestran prometedoras, pero que aún no están respaldadas por pruebas adecuadas, la cobertura con desarrollo de pruebas (CED) puede proporcionar una solución. El CED es utilizado por varios países europeos, como Bélgica, Inglaterra, Francia, Países Bajos, España y Suiza [45], y también se ha introducido en Alemania [46]. Es importante diseñar estos programas cuidadosamente, sobre todo porque las instituciones públicas a menudo no tienen experiencia en la planificación y realización de ensayos clínicos, lo que puede provocar retrasos y, dependiendo de la configuración del programa, problemas de acceso o ineficiencias [45]. Además, el CED tiene el potencial de apoyar la innovación de los pequeños y medianos fabricantes de dispositivos médicos, que de otro modo no podrían permitirse grandes ensayos clínicos.
Además, es importante dotar a los programas de CED de los mecanismos para detener el reembolso si la evidencia no es suficiente o desfavorable. En términos más generales, la implementación de enfoques de desinversión que eliminen efectivamente las tecnologías de bajo valor de la prestación de atención médica es crucial para permitir una atención óptima al paciente y una utilización óptima de los recursos, y (políticamente) difícil de lograr. Si bien las estrategias de desinversión de productos farmacéuticos en la atención ambulatoria se utilizan ampliamente en los países europeos [47], se necesitan enfoques adicionales para la desinversión de dispositivos médicos en la atención hospitalaria.
Las iniciativas destinadas a cambiar las recomendaciones de la práctica clínica, como «Choosing Wisely» y la directriz «do not do» del National Institute for Health and Care Excellence (NICE) en Inglaterra [48] podrían reducir el uso de dispositivos potencialmente ineficaces o ineficaces en función de los costes.
La eliminación de los dispositivos médicos potencialmente dañinos del mercado de la atención médica requiere mayores disposiciones de responsabilidad para los fabricantes. Con la introducción del MDR, la UE ha reforzado las disposiciones pertinentes: obliga a los fabricantes a supervisar continuamente la seguridad de los productos comercializados mediante el establecimiento de un sistema de vigilancia poscomercialización para detectar y controlar los problemas de seguridad y aplicar las medidas preventivas y correctivas necesarias (Reglamento (UE) 2017/745, art. 83) [5]. Sin embargo, la eliminación de una tecnología que ya está establecida en la práctica clínica suele enfrentarse a la resistencia de las partes interesadas relevantes [49].
Limitaciones
Este trabajo tiene varias limitaciones. Aunque la muestra de tecnologías incluidas se seleccionó con un enfoque sistemático [22], no es necesariamente representativa de todas las nuevas tecnologías. Por lo tanto, estos resultados siguen siendo indicativos.
El cálculo de la utilización se basa en la documentación del procedimiento disponible en RDC [21]. La validez de los datos está limitada por la calidad de las prácticas de codificación subyacentes, que depende tanto de la experiencia del codificador como de la medida en que los códigos utilizados sólo capturan las distintas tecnologías de la muestra (en lugar de capturar también tecnologías relacionadas para las que aún no existían códigos únicos). Además, el sistema de clasificación de procedimientos está sujeto a cambios regulares, y los códigos pueden cambiar con el tiempo a medida que la clasificación se vuelve más detallada. Esto podría haber dado lugar a una distorsión de la utilización observada para algunas tecnologías (por ejemplo, F-TUR).
Debido a la metodología de revisión rápida adoptada para identificar la evidencia, es posible que no se hayan identificado todas las citas relevantes o que se hayan excluido estudios potencialmente relevantes, por ejemplo, mediante la exclusión de idiomas distintos del alemán o el inglés, y la falta de disponibilidad de textos completos. Se eligieron fuentes adicionales para las guías clínicas y los informes de HTA desde la perspectiva del contexto alemán; Por lo tanto, estos resultados no son exhaustivos.
Por razones de viabilidad, los resultados por publicación se categorizaron basándose únicamente en las conclusiones de los autores. Esto significa que la consistencia de estas conclusiones con los resultados reportados en la sección de resultados de la publicación no se investigó más a fondo y no se ha tenido en cuenta la influencia potencial del espín [50]. Además, los estudios con resultados negativos tienen en general menos probabilidades de ser publicados [51], ya que este estudio no tuvo en cuenta el sesgo de publicación. Finalmente, diferentes autores participaron en la selección y categorización de la evidencia por tecnología. A pesar de las frecuentes reuniones y discusiones de consenso con todo el equipo de autores, no se puede excluir el sesgo del investigador.
La variable agregada que representa el conjunto de pruebas en el modelo de regresión multinivel tiene la ventaja de considerar todo el conjunto de datos disponibles hasta el momento de la utilización y evitar distorsiones, por ejemplo, debido a un único estudio negativo. Sin embargo, la desventaja es que se puede subestimar el impacto de un solo estudio crucial. Además, la consideración y ponderación de los diferentes diseños de estudio refleja una elección basada en jerarquías de evidencia y podría ser objeto de discusión. Por último, no se incorporaron al modelo otros factores observables (p. ej., financiación, advertencias de seguridad, incidencia de enfermedades) e inobservables.
Conclusiones
Este es el primer estudio que investiga la relación entre la evidencia y la utilización de una muestra de 25 nuevas tecnologías médicas en la atención hospitalaria alemana de manera descriptiva y empírica. El conjunto de pruebas clínicas por tecnología a menudo consistía principalmente en estudios no comparativos; Su robustez aumentó con el tiempo para muchas tecnologías, pero no para todas. Podría demostrarse una relación entre la evidencia y la utilización para algunas tecnologías, aunque no para todas. La influencia de la financiación y los avisos de seguridad requiere una investigación más profunda. Estos resultados revelan que podría estar justificada una reevaluación de las normas de aprobación del mercado y de las vías de ETS.
