Las bacterias gramnegativas son extraordinariamente difíciles de matar porque su membrana citoplasmática está rodeada por una membrana externa que bloquea la entrada de la mayoría de los antibióticos. La naturaleza impenetrable de la membrana externa se debe a la presencia de un gran glicolípido anfipático llamado lipopolisacárido (LPS) en su valva externa 1 . El ensamblaje de la membrana externa requiere el transporte de LPS a través de un puente proteico que se extiende desde la membrana citoplasmática hasta la superficie celular. Mantener la integridad de la membrana externa es esencial para la viabilidad de las células bacterianas y su alteración puede aumentar la susceptibilidad a otros antibióticos 2 , 3 , 4 , 5 , 6 . Por lo tanto, durante mucho tiempo se han buscado inhibidores de las siete proteínas de transporte de lipopolisacáridos (Lpt) que forman este transportador transenvoltura. Recientemente se identificó una nueva clase de antibióticos dirigidos a la máquina de transporte de LPS en Acinetobacter . Aquí, utilizando enfoques estructurales, bioquímicos y genéticos, mostramos que estos antibióticos atrapan una conformación ligada al sustrato del transportador de LPS que detiene esta máquina. Los inhibidores logran esto reconociendo un sitio de unión compuesto formado tanto por el transportador Lpt como por su sustrato LPS. En conjunto, nuestros hallazgos identifican un mecanismo inusual de inhibición del transporte de lípidos, revelan una conformación farmacológica del transportador Lpt y proporcionan la base para extender esta clase de antibióticos a otros patógenos gramnegativos.
La bacteria Acinetobacter baumannii es un retrato de la resiliencia. El microorganismo causa una variedad de infecciones y su capacidad para sobrevivir a la desecación significa que puede persistir durante semanas en los conductos de ventilación de los hospitales, los teclados de las computadoras y la piel humana. Su flexibilidad metabólica y genética le han permitido volverse resistente a los pocos antibióticos que pueden atravesar sus dos membranas celulares protectoras. Los microbios resistentes a los antibióticos matan a más de un millón de personas cada año. La amenaza global que representa A. baumannii ha colocado al microbio en un lugar destacado de la lista de patógenos prioritarios elaborada por la Organización Mundial de la Salud (OMS).
Dos estudios publicados el 3 de enero en Nature informan sobre una nueva clase de fármacos candidatos para combatir las infecciones por A. baumannii (C. Zampaloni et al. Nature https://doi.org/10.1038/s41586-023-06873-0 (2023); KP Pahil y otros, Nature https://doi.org/10.1038/s41586-023-06799-7 ; 2023). Uno de estos compuestos ya ha entrado en ensayos clínicos, pero aún está muy lejos de ser aprobado para uso clínico.Un nuevo tipo de antibiótico se dirige a una bacteria resistente a los medicamentos
Los obstáculos para desarrollar tales compuestos no son sólo científicos: los incentivos económicos son insuficientes para que muchas empresas asuman el riesgo. A medida que crece la amenaza de resistencia, la comunidad internacional debe hacer más para llevar los medicamentos prometedores desde el laboratorio hasta la cabecera de la cama.
Los nuevos compuestos bloquean la capacidad de la bacteria para transportar componentes básicos clave a donde se necesitan, y lo hacen uniéndose a un sitio nuevo en la bacteria; hay otros compuestos que se dirigen a esta vía, pero A. baumannii es resistente a ellos. Una de las moléculas, llamada zosurabalpina, mató múltiples cepas resistentes de A. baumannii en cultivo y, en ratones, una cepa resistente a todos los antimicrobianos disponibles. Se esperan los primeros resultados de los ensayos clínicos del compuesto este año.
Es raro conseguir antibióticos con un nuevo modo de acción en la clínica: sólo uno de cada 30 candidatos llega a probarse en personas. Incluso cuando un nuevo fármaco supera los ensayos clínicos y se aprueba su uso, a menudo se mantiene en reserva para los peores escenarios, por temor a que su uso generalizado acelere el día en que los microbios desarrollen resistencia a él.
Superar la fase inicial de los ensayos clínicos será sólo el comienzo. Entonces serán necesarios más estudios para evaluar el riesgo de que surja resistencia a la zosurabalpina en entornos clínicos. Luego, si se aprueba el medicamento, alguien deberá pagarlo. El mercado comercial es deprimente por diseño. Desarrollar un nuevo antibiótico suele costar más de mil millones de dólares, pero la renuencia a utilizarlo ampliamente significa que es probable que gane menos de 100 millones de dólares al año una vez en el mercado. La OMS y otros han advertido a los gobiernos sobre la magnitud del gasto, junto con la creciente amenaza de la resistencia a los antimicrobianos. Algunos gobiernos han respondido con incentivos para alentar a la industria a aceptar el desafío. Pero se ha hablado mucho más que se ha hecho.Lea el artículo: Una nueva clase de antibióticos dirigidos al transportador de lipopolisacáridos
La solución, dicen los economistas de la salud, es una combinación de incentivos para el desarrollo de nuevos fármacos antimicrobianos. Las estrategias de «empuje» están diseñadas para reducir los costos y pueden incluir más financiación gubernamental para la investigación en sus primeras etapas. Los enfoques de «atracción» recompensan a las empresas por desarrollar antibióticos exitosos; por ejemplo, los gobiernos podrían garantizar un nivel mínimo de compra, similar a las compras anticipadas realizadas de vacunas durante la pandemia de COVID-19. Los gobiernos han tendido a inclinarse más hacia estrategias de presión. Los economistas dicen que necesitan hacer más esfuerzos. Pero también deben prestar atención a las lecciones aprendidas de las vacunas contra la COVID-19, como garantizar que los precios y los contratos sean transparentes, lo que no ocurrió durante la pandemia.
El Reino Unido ha sido un líder en este sentido. En 2019, lanzó un programa de «suscripción» a través del cual las empresas reciben una tarifa anual fija que se basa en el valor de un medicamento para el sistema de atención médica, en lugar de en la cantidad de dosis vendidas. Otros países están considerando planes similares. En Estados Unidos, un equipo bipartidista de legisladores respalda la Ley de Suscripciones Antimicrobianas Pioneras para Acabar con el Aumento de la Resistencia (PASTEUR), que crearía un programa similar. Pero la ley ha tenido problemas en el Congreso de Estados Unidos desde que se introdujo por primera vez en 2020. La Unión Europea tampoco ha podido aprobar una legislación pertinente. Es hora de que los gobiernos pasen de la consideración a la acción.
Es comprensible que haya algunas dudas: el modelo de suscripción ejercería más presión sobre los presupuestos de atención médica que ya están sintiendo la presión de los precios de los medicamentos alimentados por los altos niveles de inflación. Pero hay otra manera de considerar esos pagos: como seguro contra futuras crisis sanitarias. Si los gobiernos pueden hacer viable el mercado de estos medicamentos, eso también alentará a las empresas a invertir.
En septiembre, la Asamblea General de las Naciones Unidas organizará una reunión de alto nivel para discutir la resistencia a los antimicrobianos, la primera reunión de este tipo desde 2016. Esto resaltará el problema y ofrecerá una oportunidad para obtener compromisos reales de los estados miembros. La última reunión fue transformadora en un aspecto: más de 150 países acordaron elaborar planes de acción para abordar la resistencia a los antimicrobianos.
Pero un plan de acción no es lo mismo que tomar medidas: muchos planes no se implementan ni se financian en su totalidad. Eso debe cambiar. El descubrimiento de compuestos que muestran una actividad preclínica prometedora es precisamente lo que recetó el médico. Pero sin un plan de financiación estratégico, esta receta podría permanecer en el estante durante años. Mientras tanto, A. baumannii y sus semejantes seguirán planteando una amenaza urgente para la atención sanitaria con opciones de tratamiento limitadas.
Principal
La membrana externa de las bacterias Gram-negativas es una bicapa asimétrica que contiene fosfolípidos en su valva interna y lipopolisacárido (LPS) en su valva externa 1 , 7 , 8 , 9 , 10 . La biosíntesis de LPS se completa dentro de la célula en la membrana interna. El LPS debe extraerse de la membrana interna, moverse a través del compartimento periplásmico y entregarse a través de la membrana externa a la superficie celular 1 , 9 , 11 , 12 , 13 (Fig. 1a ). Para lograr la biogénesis de la membrana externa, los componentes de la membrana interna del transportador de lipopolisacáridos, LptB 2 FGC, forman un subcomplejo que acopla la hidrólisis del ATP con la extracción de LPS de la bicapa 14 , 15 , 16 , 17 , 18 , 19 , pasándolo a la proteína. puente formado por los dominios β-jellyroll conectados de LptF, LptC, la proteína periplásmica soluble LptA y la porción periplásmica de la proteína integral de membrana LptD (refs. 6 , 14 , 20 , 21 , 22 , 23 , 24 , 25 , 26 ) . LptD, junto con su lipoproteína asociada LptE, forman el translocón de la membrana externa que sirve como conducto para que el LPS pase directamente desde el puente a la valva externa de la membrana externa 5 , 20 , 22 , 27 , 28 .
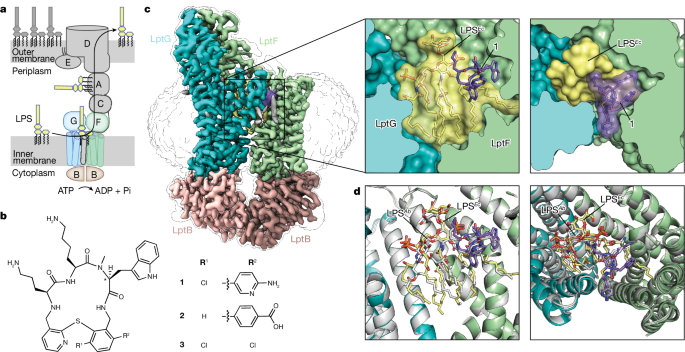
Recientemente se identificó una familia de péptidos macrocíclicos (Fig. 1b , 1 – 3 ) propuesta para apuntar a la maquinaria de transporte de lipopolisacáridos ( 1 , RO7196472; 2 , Zosurabalpin; 3 , RO7075573) 29 . Todos estos péptidos macrocíclicos tienen una actividad potente y selectiva contra cepas de Acinetobacter , incluida A. baumannii resistente a carbapenems . Un péptido macrocíclico, Zosurabalpin (compuesto 2 ), se encuentra actualmente en ensayos clínicos. Las mutaciones de resistencia al compuesto 2 se asignan a lptFG y los experimentos bioquímicos han demostrado que 2 bloquea la extracción de LPS de liposomas que contienen Acinetobacter LptB 2 FGC (ref. 29 ). Para determinar el mecanismo molecular por el cual estos antibióticos inhiben el transporte de LPS, buscamos resolver una estructura de LptB 2 FG unida a un péptido macrocíclico. Las proteínas de A. baumannii se expresaban mal y tendían a agregarse, por lo que resolvimos las estructuras de A. baylyi LptB 2 FG con los compuestos 1 a 3 a alta resolución mediante microscopía crioelectrónica (crio-EM). A. baylyi LptB 2 FG es aproximadamente un 85 % idéntico a A. baumannii (Datos ampliados, figura 1 ), es igualmente susceptible a los péptidos macrocíclicos 1 – 3 y las mutaciones que proporcionan resistencia en A. baumannii también confieren resistencia a A. baylyi (ver abajo; ref. 29 ).
El fármaco se une al LPS dentro del transportador.
Primero resolvimos una estructura de LptB 2 FG en presencia de LPS y compuesto 1 con una resolución de 3,0 Å. Inesperadamente, se descubrió que el compuesto 1 formaba contactos extensos tanto con LptB 2 FG como con una molécula de LPS unida, que es un modo único de inhibición (Fig. 1c y Datos ampliados Fig. 2 ). Debido a que LptB 2 FG se expresó heterólogamente en Escherichia coli , la estructura que obtuvimos contenía LPS de E. coli copurificado . Para descartar la posibilidad de que las diferencias estructurales entre E. coli y Acinetobacter LPS afecten la forma en que se une el LPS o cómo el compuesto 1 interactúa con el complejo LptB 2 FG-LPS, también purificamos LptB 2 FG de A. baylyi para atrapar el Acinetobacter LPS nativo ( Datos ampliados Fig. 3 ). Obtuvimos una estructura de A. baylyi LptB 2 FG con Acinetobacter LPS y 1 y solo encontramos diferencias menores en comparación con el complejo con E. coli LPS (Fig. 1d ). Los azúcares unidos al LPS de Acinetobacter se resuelven mejor y se observa una molécula de detergente ordenada en la estructura resuelta usando E. coli LPS se desplaza para acomodar la cadena lipídica adicional que está presente en Acinetobacter LPS (Datos ampliados Fig. 3g, h ). En particular, la interfaz de contacto del fármaco es casi idéntica independientemente del quimiotipo de LPS (ver más abajo) y la conformación de la proteína tampoco se modifica.
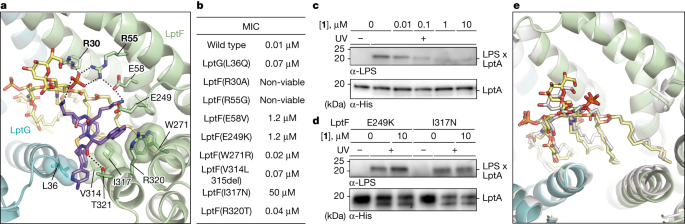
El bolsillo de unión del compuesto está revestido por cadenas laterales de varios aminoácidos en las hélices transmembrana (TM) de LptF (Glu58, Glu249, Trp271, Val314, Ile317, Arg320 y Thr321) y LptG (Leu36) (Fig. 2 ) . Experimentos previos de morbidostático llevados a cabo con el compuesto 2 identificaron mutaciones que alteraron los residuos correspondientes en A. baumannii LptFG (ref. 29 ). Para verificar que las alteraciones en estos residuos redujeron la susceptibilidad a los antibióticos peptídicos macrocíclicos, construimos cepas de A. baylyi que codifican cada variante de LptFG identificada en los experimentos de morbidistat y medimos las concentraciones inhibidoras mínimas (CMI) para los compuestos 1 a 3 contra A. baylyi . Todas las mutaciones disminuyeron la susceptibilidad de A. baylyi a los compuestos 1 , 2 y 3 (Fig. 2b y Tabla complementaria 1 ), algunas en dos o tres órdenes de magnitud. También purificamos dos complejos de A. baylyi LptB 2 FGC con sustituciones individuales de LptF (E249K o I317N) que conferían resistencia de alto nivel a los péptidos macrocíclicos (Fig. 2b ) y probamos la inhibición de la liberación de LPS en presencia de 1 (Fig. 2c ). ,d ). Debido a que el LPS de E. coli está fácilmente disponible y existen anticuerpos comerciales bien validados contra él, los experimentos bioquímicos se realizaron utilizando LPS de E. coli . El compuesto 1 bloqueó la liberación de LPS del complejo natural a LptA pero no bloqueó la liberación de LPS de ninguno de estos complejos mutantes. Estos resultados confirman la importancia de los contactos observados entre los péptidos macrocíclicos y LptFG para inhibir el crecimiento de cepas de Acinetobacter .
La droga atrapa LPS durante el transporte
La estructura LptB 2 FG –LPS– 1 sugirió que 1 atrapa un estado intermedio del transportador Lpt unido a LPS. Para evaluar si la conformación de LPS en la estructura unida al fármaco refleja un intermedio en la ruta, resolvimos estructuras de A. baylyi LptB 2 FG unidas a E. coli o Acinetobacter LPS, pero ahora en ausencia de 1 (Fig. 2e y Datos ampliados Fig. 4 ). En ambos casos, la conformación general y los contactos del LPS con el transportador son casi idénticos a los determinados en presencia del compuesto 1 (Fig. 2e ). Estos hallazgos son consistentes con un mecanismo en el que el compuesto 1 se une a un estado preexistente cargado de LPS del complejo transportador, identificando este estado como una conformación farmacológica para el desarrollo de antibióticos.
Observamos contactos de LptF (Arg30 y Arg55) con el mismo grupo fosfato en LPS que está coordinado por una amina primaria de 1 (en negrita en la Fig. 2a ). Para evaluar su importancia funcional, los mutamos por separado a Ala y Gly, respectivamente (LptF R30A y LptF R55G). Ninguna de estas variantes produjo células viables en A. baylyi , lo que sugiere que estos residuos son críticos para el plegamiento o la función de las proteínas. Descubrimos que podíamos expresar y purificar complejos con estas sustituciones de LptF, y ambas variantes de LptB 2 FG resultantes tenían actividad ATPasa comparable a la del tipo salvaje, pero ninguna transfirió LPS a LptA (Datos ampliados, figura 4 ). Por lo tanto, llegamos a la conclusión de que tanto LptF Arg30 como Arg55 son críticos para la función del complejo en Acinetobacter , probablemente porque ayudan a posicionar el LPS durante el transporte.
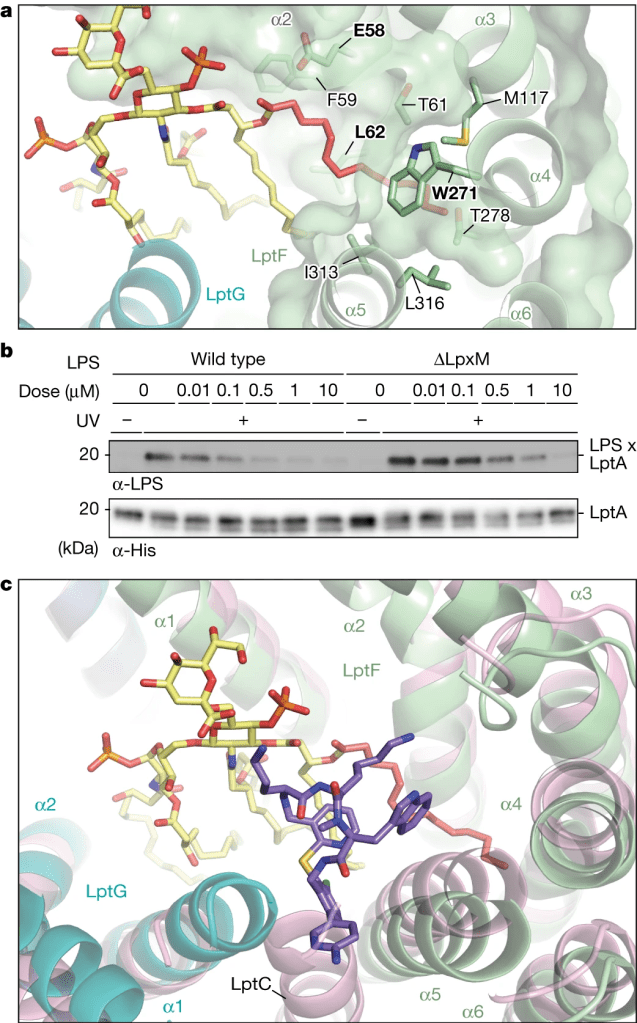
La estructura compleja ternaria también mostró una extensa interfaz entre LPS y 1 . Por lo tanto, intentamos determinar si cambiar la estructura del LPS en Acinetobacter afectaría la potencia inhibidora de 1 . La biosíntesis de LPS involucra más de 100 genes 1 , 9 , 30 , pero las mutaciones que confieren una menor susceptibilidad al compuesto fueron mutaciones de pérdida de función en lpxM (Datos ampliados, figura 5 ). LpxM realiza los pasos finales de acilación durante la biosíntesis de LPS y del área de contacto total de aproximadamente 230 Å 2 entre LPS y 1 , 94 Å 2 (41%) involucra el área de contacto entre el fármaco y la cadena de acilo instalada por LpxM (refs. 31 , 32 , 33 ) (Fig. 3a ). En una reconstitución bioquímica, el transporte de LPS aislado de una cepa de deleción LpxM de E. coli fue posible a concentraciones 20 veces mayores del compuesto 1 que las requeridas para inhibir el transporte de la estructura de LPS de tipo salvaje coincidente (Fig. 3b ). Los quimiotipos de LPS de E. coli y Acinetobacter Δ lpxM tienen patrones de acilación idénticos. De acuerdo con los experimentos bioquímicos que utilizan LPS Δ lpxM de E. coli , encontramos que la cepa de deleción lpxM de A. baylyi es 30 veces menos susceptible a 1 que la de tipo salvaje. Estudios previos en E. coli y A. baumannii han establecido que la pérdida de las cadenas de acilo graso instaladas por LpxM no impide el transporte de LPS a la membrana externa pero sí reduce la función de barrera de la membrana externa 34 , 35 . De acuerdo con esto, nuestra cepa A. baylyi ΔlpxM era hasta 1000 veces más susceptible a una amplia gama de otros antibióticos (Tabla complementaria 2 ). Debido a que se sabe que las cepas ΔlpxM tienen una virulencia reducida, la pérdida de lpxM puede no causar una susceptibilidad reducida a los péptidos macrocíclicos in vivo 35 , 36 .
El movimiento de hélice de LptC permite la unión del fármaco
LptC, un miembro del complejo Lpt de la membrana interna, contiene un dominio β-jellyroll periplásmico que desempeña un papel esencial en la transferencia de LPS de LptF a LptA (refs. 16 , 37 ). LptC también contiene una hélice TM que existe en dos estados. En uno, la hélice TM está intercalada entre las hélices TM de LptG (hélice 1) y LptF (hélices 5 y 6), que forman la puerta a través de la cual el LPS ingresa a la luz del transportador 16 , 38 , 39 . En un segundo estado, la hélice LptC se ha alejado del transportador 39 , 40 . Se cree que el movimiento de la hélice LptC entre estos estados es importante para coordinar el transporte de LPS con el ciclo catalítico de unión e hidrólisis de ATP 16 , 38 , 39 , 40 . La estructura LptB 2 FG –LPS– 1 reveló que 1 sobresale hacia la puerta formada entre la hélice 5 de LptF y la hélice 1 de LptG, lo que sugiere que su unión compite con la hélice LptC TM por la unión al complejo. Obtuvimos una estructura de A. baylyi LptB 2 FGC en presencia de LPS, aunque la densidad de la molécula de LPS se resolvió mal. En esta estructura, la hélice LptC estaba intercalada entre la hélice LptG 1 y la hélice LptF 5, similar a las estructuras determinadas previamente de otras especies 16 , 38 , 39 , 41 (Fig. 3c y Datos ampliados Fig. 6 ). A pesar de varios intentos, no pudimos obtener un complejo LptB 2 FGC al que también estuviera unido 1 . Estos datos son compatibles con un modelo en el que la unión de 1 y la unión de la hélice TM de LptC en la puerta LptFG son mutuamente excluyentes. La superposición de la estructura LptB 2 FG – LPS – 1 con la estructura LptB 2 FGC – LPS identificó cambios en la conformación de LptFG, particularmente en las hélices 4, 5 y 6 de LptF (Fig. 3 ). Esta conformación de LptF crea un bolsillo entre las hélices 4 y 5 que se adapta mejor a la cadena acilo 6 de LPS. Aunque la secuencia específica de eventos es incierta, proponemos que el movimiento de la hélice C alejándose de LptFG durante el ciclo de transporte permite que el LPS se una en el estado de transporte intermedio observado en la estructura del complejo LptB 2 FG-LPS y que este es el estado. necesaria para el reconocimiento del inhibidor.
Un modelo que requiere el movimiento de la hélice C para que se produzca la unión explicaría una observación que de otro modo sería desconcertante. Los compuestos 1 a 3 tienen una potencia celular comparable contra cepas de tipo salvaje y las mutaciones de lptFG causan reducciones similares en la susceptibilidad a los tres compuestos (Tabla complementaria 1 ). Sin embargo, cuando probamos 1 – 3 en un ensayo bioquímico que monitorea la liberación de LPS del complejo LptB 2 FGC a LptA, encontramos que el compuesto 3 era 100 veces menos efectivo que 1 y 2 (Fig. 4 ). Para identificar la base estructural de estas diferencias en el comportamiento bioquímico, determinamos las estructuras crio-EM de los compuestos 2 y 3 unidos a LptB 2 FG – LPS. Tanto 2 como 3 se unen en posiciones casi idénticas que el compuesto 1 (Fig. 4a-c y Datos ampliados Fig. 7 ). Los compuestos 1 y 2 contienen sustituyentes relativamente grandes (una aminopiridina y un benzoato de tamaño similar, respectivamente) que se superponen con la posición prevista de la hélice LptC TM en el estado de puerta ocluida (círculos discontinuos, Fig. 4a, b ). Se predice que estos sustituyentes competirán más efectivamente con la hélice LptC TM que el compuesto 3 , que contiene un átomo de cloro más pequeño en esa posición (Fig. 4c ). Si diversos grados de competencia de la hélice LptC TM son responsables de las diferentes actividades bioquímicas, 3 deberían bloquear la liberación de LPS, así como 1 y 2 cuando la hélice LptC TM está ausente (LptB 2 FGΔTM-LptC). Anteriormente se ha demostrado que ΔTM-LptC es capaz de soportar el transporte de LPS aunque carezca de la hélice TM, lo que nos permite probarlo in vitro 16 , 37 . De acuerdo con nuestra predicción, los compuestos 1 a 3 fueron igualmente efectivos para bloquear la liberación del complejo que contiene ΔTM-LptC (Fig. 4d ). Debido a que 3 es tan efectivo como 1 y 2 in vivo, llegamos a la conclusión de que los péptidos macrocíclicos se dirigen a un estado conformacional in vivo en el que la hélice C se ha movido.
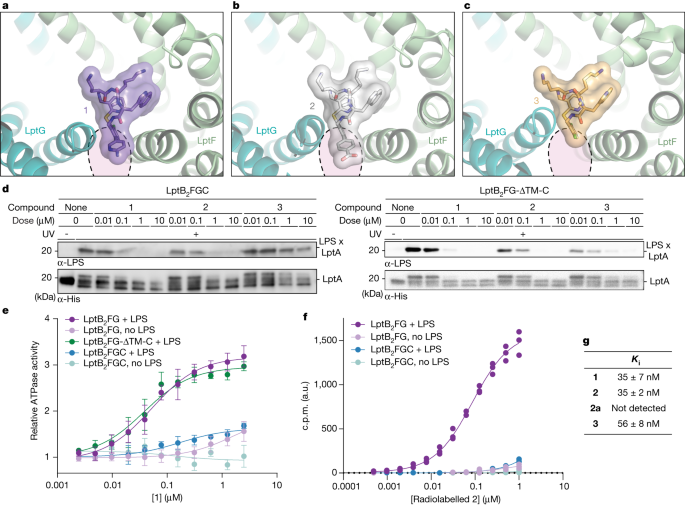
El fármaco desacopla la actividad ATPasa del transporte de LPS
Se ha demostrado que la carga de LPS en el transportador LptB 2 FG estimula la actividad ATPasa 42 . Debido a que nuestros datos han demostrado que estos péptidos macrocíclicos atrapan el LPS en el transportador, intentamos determinar si estos compuestos aumentarían la actividad de la ATPasa por encima del valor inicial unido al LPS. La adición de los compuestos 1 a 3 a los complejos LptB 2 FG y LptB 2 FGΔTM-LptC condujo a un gran aumento en la actividad de la ATPasa (Fig. 4e y Datos ampliados Fig. 8 ). Esta actividad dependía de la presencia de LPS, lo que respalda la importancia del LPS para la unión del fármaco al transportador. Los resultados de los ensayos de actividad ATPasa y liberación de LPS sugirieron que el mejor indicador de la actividad in vivo de los péptidos macrocíclicos es la afinidad de un compuesto por LptB 2 FG, en ausencia de LptC. De hecho, un ensayo de centelleo de proximidad (SPA) mostró que el 2 radiomarcado se unía a LptB 2 FG pero no a LptB 2 FGC y esa unión dependía de la presencia de LPS (Fig. 4f ). De acuerdo con su potencia similar en ensayos celulares, 1 , 2 y 3 tenían capacidades similares para desplazar competitivamente H 3 – 2 de LptB 2 FG ( K i = 35, 35 y 56 nM, respectivamente). Por lo tanto, la ATPasa y los ensayos de unión con complejos que carecen de LptC o su hélice TM recapitularon los hallazgos in vivo de que 1 – 3 tienen actividad comparable, lo que sugiere nuevamente que los péptidos macrocíclicos descritos aquí apuntan a un estado en el que la hélice LptC TM se ha disociado del complejo.
Hemos demostrado que una nueva familia de antibióticos peptídicos macrocíclicos mata a Acinetobacter atrapando LPS mientras este sustrato está en tránsito dentro del transportador de lipopolisacáridos. Debido a que Acinetobacter no requiere LPS para la viabilidad , se justifica un comentario aquí sobre el mecanismo de muerte celular. Trabajos anteriores han demostrado que muchos genes implicados en la biogénesis de LPS en Acinetobacter son condicionalmente esenciales y sólo pueden eliminarse si se bloquea el inicio de la biosíntesis de LPS 43 , 44 . La acumulación tóxica de intermediarios de la biosíntesis de LPS se produce cuando se inicia el transporte de LPS pero no puede completarse. Descubrimos que las cepas de A. baylyi que carecen de LPS (Δ lpxC ) pueden crecer in vitro en presencia de concentraciones muy altas de 1 (Tabla complementaria 3 ). Por lo tanto, el fármaco no actúa agotando el LPS de la membrana externa porque estas células pueden vivir sin LPS en la membrana externa, sino a través de su acumulación tóxica dentro de la célula. Aunque la pérdida de LPS en Acinetobacter proporciona un mecanismo para escapar de la susceptibilidad a los medicamentos, disminuye significativamente tanto la aptitud como la virulencia 43 , 45 , 46 . Queda por ver si la eliminación de LPS es una estrategia viable para reducir la susceptibilidad al tratamiento con péptidos macrocíclicos in vivo.
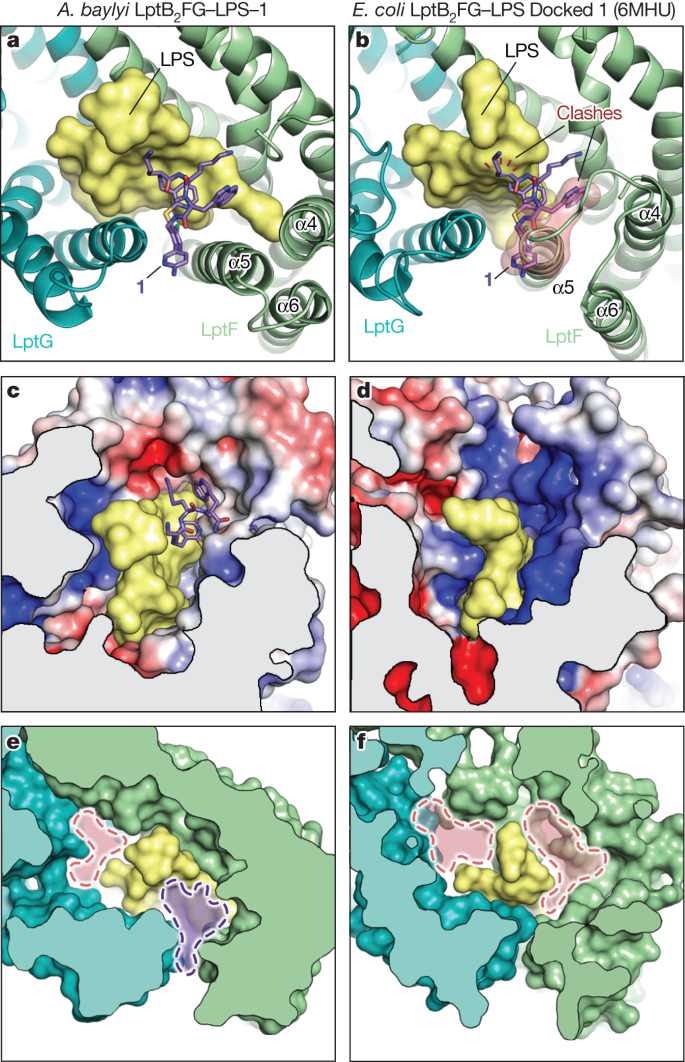
Los inhibidores de péptidos macrocíclicos son muy potentes contra cepas de Acinetobacter pero son inactivos contra otros organismos Gram-negativos y nos hemos preguntado qué lecciones contienen nuestras estructuras y experimentos mecanicistas para comprender esta susceptibilidad estrecha a los medicamentos. Hemos demostrado que la capacidad de los péptidos macrocíclicos para unirse a Acinetobacter LptB 2 FG requiere LPS, pero las estructuras revelan que estos inhibidores contactan solo con las regiones más conservadas del núcleo del lípido A del LPS. Por lo tanto, la variación de la estructura del LPS por sí sola no explica la selectividad de especies de estos medicamentos (Fig. 1d ). La pose del fármaco observada en las estructuras ternarias de A. baylyi LptB 2 FG en complejo con LPS de E. coli explicó completamente las mutaciones de resistencia que se aislaron en Acinetobacter , lo que indica que los contactos con LptFG son críticos para el reconocimiento del fármaco. Además, los resultados de los ensayos de transporte bioquímico utilizando LPS de E. coli fueron consistentes con los ensayos celulares que se realizaron en cepas de Acinetobacter . Por lo tanto, parece probable que la selectividad de especies se deba a diferencias en las proteínas Lpt. Las proteínas bacterianas homólogas de diferentes géneros suelen tener una baja conservación de secuencia. Aunque las secuencias de A. baumannii y A. baylyi LptFG son idénticas en un 82 % y comparten casi todos los residuos (16 de 18) que entran en contacto con LPS o con el péptido macrocíclico, las proteínas LptFG de E. coli son sólo un 25 % idénticas a sus homólogas de Acinetobacter . y la mayoría de los residuos que entran en contacto con LPS son diferentes (Datos ampliados, Fig. 1 ). Las estructuras de LptB 2 FG de E. coli determinadas previamente en complejo con LPS de E. coli muestran que el LPS ocupa una posición diferente en la cavidad central de LptFG (refs. 38 , 39 ) (Fig. 5 ). Además, existen diferencias en las posiciones de algunas de las hélices de LptF que darían como resultado un choque con los péptidos macrocíclicos, así como diferencias en la superficie electrostática que rodea las bolsas de unión de LptB 2 FG-LPS (Fig. 5c, d ). De hecho, el FGC purificado de E. coli LptB 2 es más de 1000 veces más resistente a la inhibición por parte del candidato clínico, 2 , que su homólogo de Acinetobacter (Fig. 2c y Datos ampliados, Fig. 8d) .). Por lo tanto, el espectro estrecho refleja las diferencias en las proteínas, que afectan la forma en que se unen al LPS. Observamos que existen otras bolsas de unión que rodean al LPS en E. coli LptB 2 FG y, por lo tanto, puede ser posible diseñar inhibidores análogos para este u otros patógenos gramnegativos que atrapan un estado intermedio de unión a LPS (Fig. 5e, f ) . . En términos más generales, el mecanismo de estos pegamentos moleculares proporciona una hoja de ruta para el desarrollo de otros compuestos que se unen a un transportador y su sustrato simultáneamente para bloquear el transporte de lípidos en sistemas procarióticos y eucarióticos 47 .
Métodos
No se utilizaron métodos estadísticos para predeterminar el tamaño de la muestra. Los experimentos no fueron aleatorios y los investigadores no estaban cegados a la asignación durante los experimentos ni a la evaluación de resultados.
SDS-PAGE e inmunotransferencia
Se utilizaron geles de gradiente de poliacrilamida de 4 a 20 % de Tris-HCl caseros o geles de proteína prefabricados Mini-PROTEAN TGX de 4 a 20 % (Bio-Rad) con tampón de ejecución Tris-glicina. El tampón de carga de muestra 2X SDS se refiere a una mezcla que contiene Tris 125 mM (pH 6,8), 4 % (p/v) de SDS, 30 % (v/v) de glicerol, 0,005 % de azul de bromofenol y 5 % (v/v) de glicerol. β-mercaptoetanol. Los geles de electroforesis en gel de SDS-poliacrilamida (SDS-PAGE) se ejecutaron durante 45 a 60 minutos a 200 V. Los complejos de proteínas purificados para crio-EM se analizaron mediante SDS-PAGE, seguido de tinción con azul de Coomassie (Alfa Aesar) y obtención de imágenes utilizando el Gel. característica de un generador de imágenes Azure Biosystems C400. Para la transferencia Western, las proteínas se transfirieron a membranas de PVDF Immun-Blot (Bio-Rad). Luego se bloquearon las membranas utilizando tampón de bloqueo de caseína filtrado esterilizado (Sigma-Aldrich) durante 1 h y posteriormente se incubaron con los anticuerpos apropiados. Se utilizaron los siguientes anticuerpos primarios: conjugado de ratón anti-His HRP (Biolegend, 652504, dilución 1:10.000) y monoclonal de ratón anti-LPS core (Hycult Biotechnology, HM6011, clon WN1 222-5, lote nº 18419M0715-A, 1 : dilución 5.000). Se utilizaron los siguientes anticuerpos secundarios: conjugado RP burro-anti-conejo (GE Amersham, NA934-1ML, lote n.º 16801031, dilución 1:10.000), conjugado HRP oveja-anti-ratón (GE Amersham, LNA931V/AH, lote n.º .14251045, dilución 1:10.000). Las bandas se visualizaron utilizando el reactivo de detección de transferencia Western ECL Prime (GE Amersham) y un sistema de imágenes Azure c400. Las inmunotransferencias sin recortar están disponibles en la figura complementaria 1 .
Plásmidos, cepas y oligonucleótidos.
Los genes que codifican LptB, LptC y LptFG se amplificaron mediante reacción en cadena de la polimerasa (PCR) a partir del ADN genómico de Acinetobacter baylyi ADP1 (ATCC 33305). Los productos de PCR lptB y lptFG se insertaron en pCDFduet mediante ensamblaje Gibson (New England Biolabs) para generar plásmidos análogos a los utilizados para otros homólogos de LptB 2 FG 16 . Se usó un diseño similar para el plásmido modificado pTRAB-FLAG-LptB-LptFG para la purificación del mismo complejo del huésped nativo, que se construyó combinando los amplicones de ADNg de los mismos marcos de lectura abiertos incorporando una etiqueta FLAG sin enlazador en el extremo N. término de LptB, un promotor trp modificado de E. coli y regiones adyacentes de pTRC99a, un replicón híbrido pBR322WH1266 y un casete de resistencia a espectinomicina de pCDFduet mediante ensamblaje Gibson. Se añadió una etiqueta His 7 N-terminal sin conector a LptB en pCDFduet utilizando el ensamblaje de ADN NEBuilder HiFi (New England Biolabs). Los productos de PCR de lptC se insertaron en pET22/42 con un sitio de escisión de trombina C-terminal y una etiqueta His 7 . Los cebadores de oligonucleótidos se adquirieron de Eton Biosciences, Genewiz o Integrated DNA Technologies. Los plásmidos y cepas utilizados en este estudio se informan en las Tablas complementarias 4 y 5 , respectivamente. Las secuencias de plásmidos se encuentran a continuación.
Construcción y uso de cepas mutantes de A. baylyi.
El cultivo, la manipulación genética y las mediciones de CIM de A. baylyi ADP1 se realizaron de acuerdo con los procedimientos informados anteriormente 43 , 48 . Los mutantes puntuales se construyeron en un procedimiento de dos pasos siguiendo la ref. 49 con la introducción y escisión del casete de integración en el codón 66 de pepA , en el que el fragmento de escisión de la secuencia de ADN cromosómico de tipo salvaje desde el codón 406 de pepA hasta el codón 193 de lptG portaba la mutación deseada y los clones resultantes se cribaron mediante amplicón. secuenciación del codón 81 de HolC al codón 501 de GpmI , mientras que la eliminación de lpxM se logró siguiendo el mismo procedimiento, excepto que la inserción y escisión del casete de integración eliminó los codones 79-279 de lpxM para evitar la interferencia con genes vecinos y superpuestos que una eliminación mayor puede poner en riesgo. y reemplazó el codón 78 con un codón de parada ocre para evitar una lectura completa que dé como resultado una fusión aberrante, con la secuencia del fragmento de escisión que abarca desde el codón 211 de sppA hasta el codón 497 de ComA y verificada mediante secuenciación de amplicones desde el codón 94 de MhpC hasta el codón 327 de ComA. así como por ausencia de un producto de PCR correspondiente a una región que abarca los codones 79 a 279 de lpxM para comprobar si hay duplicaciones. De la misma manera se realizó una deleción de la proteasa lon para producir la tinción utilizada para la expresión y purificación de LptB 2 FG para imitar la cepa BL21 de E. coli utilizada en el resto de purificaciones, para lo cual la región que abarca 72 pares de bases ( pb) corriente arriba del codón de inicio lon y 1 pb corriente abajo del codón de parada lon se escindieron después de ser reemplazados con el mismo casete de integración, produciendo una eliminación sin marcadores, con la secuencia del fragmento de escisión abarcando desde el codón 491 de ArnT hasta el codón 40 de 45_DOPA_Dioxygenase y verificado mediante secuenciación de amplicones que abarca desde el codón 322 de ArnT hasta el codón 221 de 45_DOPA_Dioxygenase así como por la ausencia de un producto de PCR correspondiente a una región que abarca desde los codones 328 a 768 de lonpara comprobar si hay duplicaciones. Después de la confirmación del amplicón, se analizó la susceptibilidad de tres aislados validados de cada mutante construido a un panel de antibióticos con antibióticos conocidos con mecanismos de acción conocidos como un paso de validación adicional para garantizar la congruencia de los fenotipos entre las réplicas, lo cual se confirmó en todos los casos y en uno de los casos. Las réplicas validadas se utilizaron posteriormente para las mediciones de MIC que se informan aquí. En el caso de R30A y R55G, no se pudo aislar ninguna colonia que incorporara estas mutaciones, mientras que el enfoque idéntico introdujo fácilmente sustituciones conservadoras de R30K y R55K, lo que resultó en una mayor sensibilidad a los antibióticos a pesar de su naturaleza leve, lo que indica que los deterioros causados por la sustitución de diferentes los residuos en esas posiciones no sobreviven.
determinación de CMI
Las determinaciones de CIM se realizaron mediante microdilución en caldo de acuerdo con las pautas del CLSI (CLSI M07-A11 2018). Los inóculos bacterianos se prepararon diluyendo cultivos líquidos durante la noche en LB. Se inocularon paneles antibacterianos que contenían soluciones antibacterianas con un volumen apropiado de inóculo para dar un inóculo final de aproximadamente 5 × 10 5 ufc ml -1 y las concentraciones de prueba deseadas de agentes antibacterianos en placas estándar de 96 pocillos con 0,1 ml de cultivo por pocillo. Las placas de prueba se incubaron durante 20 a 24 h y se registró la densidad óptica (DO 600 ) utilizando un lector de placas. Los valores de MIC correspondieron a la concentración más baja del compuesto que inhibe el crecimiento bacteriano más allá de la cual la OD dejó de disminuir.
Purificación de complejos LptB 2 FG para crio-EM
Los complejos LptB 2 FG se purificaron como se describió anteriormente, con ligeras modificaciones 17 . Se diluyeron cultivos nocturnos de Bl21(λDE3) E. coli que contenían pCDFduet-His 7 LptB-LptFG o A. baylyi que contenía pTRAB-FLAGLptB-LptFG 1:100 en LB o caldo excelente que contenía 50 mg l- 1 de espectinomicina. Las células se cultivaron a 37 °C (o 30 °C para A. baylyi ) hasta una DO 600 de aproximadamente 0,8. Luego se agregaron IPTG 200 µM y glucosa al 0,2% (o IPTG 500 µM para A. baylyi ) y se permitió que las células crecieran durante otras 2 a 3 h. Las células se recogieron mediante centrifugación (4200 g , 20 min, 4 °C). Los sedimentos celulares se congelaron instantáneamente con nitrógeno líquido y se almacenaron a -80 °C. Todos los pasos posteriores se llevaron a cabo a 4 °C a menos que se indique lo contrario.
Los sedimentos celulares descongelados se resuspendieron en tampón de lisis (Tris 50 mM (pH 7,4), NaCl 300 mM, PMSF 1 mM, 100 μg ml −1 de lisozima, 50 μg ml −1 de DNasa I, 1 tableta completa de cóctel inhibidor de proteasa por 40 ml) homogeneizados y sometidos a paso a través de un disruptor celular de alta presión EmulsiFlex-C3 tres veces. El lisado celular se centrifugó (10.000 g , 10 min) y el sobrenadante se centrifugó adicionalmente (100.000 g , 1 h). Los sedimentos resultantes se resuspendieron y se solubilizaron en tampón de solubilización (Tris 20 mM (pH 7,4), NaCl 300 mM, glicerol al 15 %, MgCl2 5 mM, DDM al 1 % (peso/vol) (Anatrace Maumee), PMSF 100 μM, 2 mM. ATP) y se agitó a 4 °C durante 2 h. ( El lisado de células de A. baylyi se sometió inmediatamente a solubilización con detergente sin los pasos de centrifugación anteriores ni adición de ATP, pero se usó 0,35 µM 1 para complementar algunos lotes desde el paso de solubilización en adelante). La mezcla se centrifugó (100.000 g , 30 min), el sobrenadante se añadió con imidazol hasta una concentración final de 15 mM y luego se agitó con resina Ni-NTA Superflow (Qiagen) durante 1 h. ( El sobrenadante de A. baylyi también se filtró a través de una membrana PVDF Durapore (Millipore-Sigma) con un tamaño de poro de 0,45 µM y se incubó con resina de agarosa M2-FLAG (Millipore-Sigma) sin suplemento de imidazol en lugar de resina Ni-NTA Superflow). Luego, la resina se lavó con tampón de afinidad de 2 x 10 volúmenes de columna (NaCl 300 mM, Tris 20 mM (pH 7,4), glicerol al 15 %, DDM al 0,01 % (peso/vol), GDN al 0,04 % (peso/vol) (Anatrace Maumee )) que contiene imidazol 20 mM seguido de 2 × 15 volúmenes de columna de tampón de afinidad que contiene imidazol 35 mM. ( Los lotes derivados de A. baylyi se lavaron con 3 x 10 volúmenes de columna de tampón de afinidad). La proteína se eluyó con 2 x 2 volúmenes de columna de tampón de afinidad que contenía imidazol 200 mM (12,5 volúmenes de columna de tampón de afinidad suplementados con 0,2 mg ml – 1 de péptido FLAG (Genscript) para lotes derivados de A. baylyi ) concentrado utilizando un microscopio molecular de 100 kDa. corte de peso en filtro centrífugo Amicon Ultra (Millipore) y purificado mediante cromatografía de exclusión por tamaño en una columna de aumento Superdex 200 en tampón SEC (NaCl 300 mM, Tris 20 mM (pH 7,4), GDN al 0,02 %, tris(hidroxipropil)fosfina 0,25 mM) . Las fracciones se combinaron y se concentraron a 7–8 mg ml −1 usando un filtro centrífugo Amicon Ultra de corte de peso molecular de 100 kDa. Luego se preparó la proteína para microscopía como se describe a continuación.
Purificación de complejos LptB 2 FGC para crio-EM
La purificación se realizó en gran medida como se describe para LptB 2 FG con las siguientes modificaciones. La expresión se realizó utilizando E. coli C43(λDE3) que contenía pCDFduet-LptB-LptFG y pET22/42-LptC-trombina-His 7 . Los cultivos se cultivaron en presencia de 50 mg l -1 de espectinomicina y 50 mg l -1 de carbenicilina. El resto de la expresión y purificación se realizó de manera idéntica a la purificación de LptB 2 FG hasta el paso de cromatografía de exclusión por tamaño. Las fracciones recogidas después de la cromatografía de exclusión por tamaño se incubaron durante la noche con trombina de grado de restricción (Sigma) para escindir la etiqueta His. La solución se añadió con imidazol 8 mM y la proteína no escindida se eliminó pasándola a través de resina Ni-NTA y benzamidina Sefarosa. Las fracciones se combinaron y se concentraron a 7–8 mg ml −1 usando un filtro centrífugo Amicon Ultra de corte de peso molecular de 100 kDa. Luego se preparó la proteína para microscopía como se describe a continuación.
Recopilación de datos de microscopía electrónica.
La proteína se purificó como se describió anteriormente y luego se incubó en hielo con 0,2 mg ml -1 de lipopolisacáridos de E. coli EH100 (mutante Ra; Sigma-Aldrich) y fármaco 0,25 mM (si corresponde) durante 45 minutos con agitación suave. Para las proteínas purificadas de Acinetobacter , no se agregaron lipopolisacáridos de E. coli . Luego se aplicó la muestra a rejillas de cobre de malla 400, carbono con orificios de 20 nm, plano C, descargado incandescente, con un diámetro de orificio de 1,2 μm, una separación entre orificios de 1,3 μm y una malla de 400 (Protochips). Las rejillas se secaron durante 6,5 s a 4 °C y 100% de humedad con la fuerza de transferencia establecida en 12 y se congelaron instantáneamente con etano líquido enfriado con nitrógeno usando un Thermo Fisher Scientific Vitrobot Mark IV (Thermo Fisher Scientific). Luego, la rejilla se cargó en un criomicroscopio electrónico Titan Krios G3i (Thermo Fisher) operado a un voltaje de aceleración de 300 kV. Las pilas de imágenes (videos) se grabaron en un filtro de imágenes Gatan Bioquantum K3 (Gatan), usando el modo de conteo y una ampliación calibrada de × 105,000 y un tamaño de píxel de 0.825 Å, usando SerialEM 50 . La rendija del filtro de energía se ajustó a 20 eV con un rango de desenfoque de entre 1,1 y 2,2 μm. El tiempo de subtrama se configuró para permitir la recopilación de 50 subtramas por pila de imágenes con una tasa de dosis de electrones de aproximadamente 1 e – por Å 2 por cuadro. La dosis total de electrones fue de aproximadamente 50 e – por Å 2 . Se utilizó el esquema multidisparo en SerialEM para la recopilación de datos, con configuraciones de nueve hoyos por movimiento de etapa y dos disparos por hoyo. La recolección de datos para todas las estructuras se realizó de la misma manera.
Procesamiento de imágenes y reconstrucción tridimensional.
Los fotogramas de vídeo se corrigieron por movimiento y se ponderaron por dosis y los parámetros de la función de transferencia de contraste (CTF) se estimaron utilizando CryoSPARC Live 51 . La recolección de partículas se llevó a cabo utilizando el selector de gotas crioSPARC y las partículas basura se filtraron mediante rondas sucesivas de clasificación bidimensional en crioSPARC. Los modelos iniciales se generaron utilizando la reconstrucción ab initio en crioSPARC y luego las partículas se filtraron mediante rondas sucesivas de refinamiento heterogéneo. Después de un trabajo inicial de refinamiento no uniforme, las partículas fueron sujetas a corrección de movimiento local, estimación de CTF de parche, refinamiento de CTF local y refinamiento de CTF global (apto para inclinación del haz, trébol del haz y aberración esférica). Luego, las partículas fueron sometidas a un refinamiento no uniforme para producir la reconstrucción global final. Los mapas se refinaron aún más mediante resta de partículas y refinamiento local con una máscara centrada en la TM y los dominios de unión de nucleótidos del transportador. Para todos los mapas, también probamos la clasificación sin alineación en Relion. En el mejor de los casos, esto solo produjo mejoras nominales en la resolución después de volver a importarlos a crioSPARC y realizar un refinamiento no uniforme en comparación con los mapas de preclasificación. La clasificación 3D sin alineamientos en crioSPARC reveló varias conformaciones posibles del fármaco dentro del transportador, como se destaca en los datos ampliados Fig. 3b,f 52 , 53 . Los mapas utilizados para las figuras se filtraron según la resolución local con factor B de nitidez dentro de cryoSPARC o mediante posprocesamiento realizado en DeepEMhancer 54 . Las aplicaciones de biología estructural utilizadas en este proyecto fueron compiladas y configuradas por SBGrid 55 .
Construcción, refinamiento y validación de modelos.
Los modelos iniciales para LptB, LptF y LptG se generaron utilizando SwissModel 56 . Las estructuras resultantes se acoplaron al mapa LptBFG utilizando Chimera 57 . Las restricciones Cif para lipopolisacárido de E. coli se generaron utilizando la herramienta de dibujo en CCP4 (ref. 58 ). Las restricciones Cif para lipopolisacárido de Acinetobacter se generaron utilizando el servidor web Grade2 de Global Phasing Limited. Se generaron restricciones Cif para los péptidos macrocíclicos utilizando eLBOW 59 . Luego, las coordenadas se refinaron usando Phenix 60 , 61 . El modelo se optimizó aún más utilizando ISOLDE 62 , al que se accede a través de ChimeraX 63 . La construcción manual de modelos se llevó a cabo en Coot 64 . El modelo final se inspeccionó visualmente para determinar el ajuste general al mapa y se inspeccionó adicionalmente utilizando MolProbity y la estimación de calidad local de residuos DAQ 65 , 66 . Todos los residuos en nuestros modelos tenían puntuaciones DAQ> 0, excepto aquellos contenidos en la hélice de LptC. La hélice de LptC se modela como polialanina porque nuestros mapas no eran de calidad suficiente para permitir una asignación inequívoca del registro de hélice. Las estadísticas de validación del modelo se resumen en la Tabla 1 de datos ampliados .
Purificación de complejos LptB 2 FG para reconstitución bioquímica.
LptB 2 FG utilizado para experimentos bioquímicos se purificó como se describe para crio-EM con las siguientes modificaciones. El tampón de afinidad fue NaCl 300 mM, Tris 20 mM (pH 7,4), glicerol al 10 %, DDM al 0,015 % (peso/vol). El tampón SEC fue NaCl 300 mM, Tris 20 mM (pH 7,4), glicerol al 5 %, DDM al 0,05 % y tris(hidroxipropil)fosfina 0,5 mM.
Purificación de complejos LptB 2 FGC para reconstitución bioquímica.
LptB 2 FGC utilizado para experimentos bioquímicos se purificó como se describe para crio-EM con las siguientes modificaciones. El tampón de afinidad fue NaCl 300 mM, Tris 20 mM (pH 7,4), glicerol al 10 %, DDM al 0,015 % (peso/vol). El tampón SEC fue NaCl 300 mM, Tris 20 mM (pH 7,4), glicerol al 5 %, DDM al 0,05 % y tris(hidroxipropil)fosfina 0,5 mM. Los complejos de E. coli LptB 2 FGC se purificaron como se describió anteriormente [ 17] .
Purificación de LptA I36 p BPA
LptA I36 p BPA se purificó como se describió anteriormente [17] . Brevemente, se cultivaron células Bl21 (λDE3) de E. coli que contenían pSup-BpaRS-6TRN y pET22b-LptA(I36Am) hasta una DO 600 de aproximadamente 0,6 a 37 °C en medio LB que contenía 50 μg ml -1 de carbenicilina, 30 μg ml −1 de cloranfenicol y pBPA 0,8 mM (BaChem). Luego se indujeron las células con IPTG 50 µM; se dejó crecer durante 2 h; cosechado; resuspendido en una mezcla que contenía Tris-HCl 50 mM (pH 7,4), sacarosa 250 mM y EDTA 3 mM; se incubó en hielo durante 30 min; y granulado (6.000 g , 10 min). El sobrenadante se complementó con PMSF 1 mM e imidazol 10 mM y se sedimentó (100.000 g , 30 min). El sobrenadante se incubó con resina Ni-NTA, que luego se lavó dos veces (20 volúmenes de columna de Tris-HCl 20 mM (pH 8,0), NaCl 150 mM, glicerol al 10 % (vol/vol) e imidazol 20 mM). La LptA se eluyó dos veces (2,5 volúmenes de columna de tampón de lavado suplementados con otro imidazol 180 mM), se concentró utilizando un concentrador centrífugo Amicon de corte de 10 kDa (Millipore), se congeló instantáneamente y se almacenó a -80 °C hasta su uso.
Preparación de liposomas LptB 2 FG o LptB 2 FGC
Los proteoliposomas se prepararon como se describió anteriormente [17] . Se sonicaron brevemente extracto lipídico polar acuoso de E. coli (Avanti Polar Lipids) (30 mg ml -1 ) y LPS acuoso de E. coli EH100 (mutante Ra; Sigma) (2 mg ml -1 ) para la homogeneización. Para los experimentos que prueban el efecto de la estructura del LPS, utilizamos LPS aislado de GKM374 (BL21DE3 eptA :: catR arnA :: kanR eptC :: gentR ) o TXM418 (BL21DE3 eptA :: catR arnA :: FRT eptB :: gentR lpxM :: kanR ) como se describió anteriormente 33 . Se preparó una mezcla de Tris-HCl 20 mM (pH 8,0), NaCl 150 mM, 7,5 mg ml -1 de lípidos polares de E. coli , 0,5 mg ml -1 de LPS y DDM al 0,25% y se mantuvo en hielo durante 10 min. Se añadió LptB 2 FGC o LptB 2 FG purificada hasta una concentración final de 0,86 µM y la mezcla se dejó en hielo durante 20 minutos. La mezcla se diluyó 100 veces con Tris-HCl 20 mM frío (pH 8,0) y NaCl 150 mM y se mantuvo en hielo durante 20 minutos. Los proteoliposomas se sedimentaron (300.000 g , 2 h, 4 °C), se resuspendieron en Tris-HCl 20 mM (pH 8,0) y NaCl 150 mM, se diluyeron 100x y se centrifugaron (300.000 g , 2 h, 4 °C). Los sedimentos se resuspendieron en una mezcla de Tris-HCl 20 mM (pH 8,0), NaCl 150 mM y glicerol al 10% (250 µl por 100 µl de la solución de predilución original), se homogeneizaron mediante sonicación, se congelaron instantáneamente y se almacenaron a -80°. C hasta su uso.
Purificación de LptC(ΔTM)
LptC(ΔTM) se purificó en gran medida como se describió anteriormente [16] . Brevemente, se cultivaron células Bl21 (λDE3) de E. coli que contenían pET22/42-LptC(ΔTM)-His 7 hasta una DO 600 de aproximadamente 0,6 a 37 °C en medio LB que contenía 50 μg ml -1 de carbenicilina. Luego se indujeron las células con IPTG 50 µM; se dejó crecer durante 2 h; Se recogieron y se resuspendieron en tampón de lisis (Tris 50 mM, pH 7,4, NaCl 300 mM, EDTA 0,1 mM). Se añadieron lisozima, DNasaI y PMSF hasta concentraciones finales de 100 µg ml -1 , 50 µg ml -1 y 1 mM, respectivamente. Las células se homogeneizaron y se sometieron a paso tres veces a través de un disruptor celular de alta presión EmulsiFlex-C3. El lisado celular se centrifugó (10.000 g , 10 min) y el sobrenadante se centrifugó adicionalmente (100.000 g , 1 h). Al sobrenadante se le añadió imidazol hasta una concentración final de 15 mM y luego se agitó con resina Ni-NTA Superflow (Qiagen) durante 1 h. Luego, la resina se lavó con 2 x 10 volúmenes de columna de tampón de afinidad (NaCl 300 mM, Tris 20 mM (pH 7,4), glicerol al 15%) que contenía imidazol 20 mM, seguido de 2 x 15 volúmenes de columna de tampón de afinidad que contenía imidazol 35 mM. La proteína se eluyó con 2 x 2 volúmenes de columna de tampón de afinidad que contenía imidazol 200 mM, se concentró utilizando un filtro centrífugo Amicon Ultra (Millipore) de corte de peso molecular de 10 kDa y se purificó mediante cromatografía de exclusión por tamaño en una columna de aumento Superdex 200 en tampón SEC (300 NaCl mM, Tris 20 mM (pH 7,4), glicerol al 5%). Las fracciones se combinaron y almacenaron a -80 °C.
Ensayo de liberación de LPS
Las cantidades de liberación de LPS de los proteoliposomas a LptA se midieron como se describió anteriormente 17 . Los ensayos utilizaron proteoliposomas al 60 % (en volumen) en una solución que contenía Tris-HCl 50 mM (pH 8,0), NaCl 500 mM, glicerol al 10 % y LptA I36 p BPA 2 µM . Las mezclas de reacción se incubaron con el fármaco durante 10 minutos a temperatura ambiente, según correspondiera. Luego se iniciaron las reacciones mediante la adición de ATP y MgCl 2 (concentraciones finales de 5 mM y 2 mM, respectivamente) y se llevaron a cabo a 30 °C. Se retiraron alícuotas (25 µl) de las mezclas de reacción y se irradiaron con luz ultravioleta (UV) (365 nm) en hielo durante 10 minutos usando una lámpara B-100AP (Fisher Scientific). Después de la irradiación UV, se agregaron 25 µl de tampón de carga de muestra SDS-PAGE 2 ×, las muestras se hirvieron durante 10 minutos y las proteínas se separaron usando geles de gradiente de poliacrilamida Tris-HCl al 4-20% con tampón de funcionamiento Tris-glicina. La inmunotransferencia se realizó como se describe anteriormente.
Ensayo de ATPasa
Los ensayos de ATPasa se realizaron utilizando un método de molibdato modificado, como se informó anteriormente, con ligeras modificaciones 17 . Los ensayos utilizaron proteoliposomas al 30% (en volumen) en una mezcla que contenía Tris-HCl 50 mM (pH 8,0), NaCl 500 mM, glicerol al 10% y MgCl2 2 mM . La mezcla de reacción que contenía proteoliposoma se incubó con el fármaco a temperatura ambiente durante 10 minutos, según correspondiera. Las reacciones se iniciaron mediante la adición de ATP hasta una concentración final de 5 mM y se ejecutaron a 30 °C. Se tomaron alícuotas (5 µl) a los 0, 15, 30 y 45 min. Las reacciones se apagaron con un volumen igual de SDS al 12%. Las cantidades de Pi se determinaron mediante un método colorimétrico y se utilizó fosfato de potasio como estándar 43 . Los reactivos se obtuvieron de Sigma-Aldrich. Después de la adición de SDS, se añadió una mezcla que contenía 10 µl de 30 mg ml -1 de ácido ascórbico, HCl 0,5 N, 5 mg ml -1 de molibdato de amonio y SDS al 6%. Las muestras se incubaron a temperatura ambiente durante 7 min y se agregaron 15 µl de una solución acuosa que contenía 20 mg ml −1 de citrato de sodio tribásico dihidrato, 2 mg ml −1 de arsenito de sodio y ácido acético al 2% (vol / vol). La absorbancia a 850 nm se midió usando un Spectramax Plus 384 (Molecular Devices) después de 20 min. Las barras de error indican las desviaciones estándar de las tasas promedio medidas en tres réplicas biológicas.
Ensayo de centelleo-proximidad
La unión del radioligando [ 3H ]-RO7223280 a abl LptB 2 FG y abl LptB 2 FGC se midió mediante SPA basado en perlas. Todos los pasos se realizaron en tampón SEC (Tris 20 mM, pH 7,5, NaCl 300 mM, glicerol al 5 %, TCEP 0,5 mM, DDM al 0,05 % ± 10 µM de E. coli J5 LPS(Rc) TLRGRADE (Enzo Life Sciences)) y a 4 °C a menos que se indique lo contrario. La proteína purificada se incubó primero con perlas de etiqueta HIS de PVT de cobre (Perkin Elmer) durante 1,5 h con una rotación suave. Se agregaron doce concentraciones de radioligando a la mezcla PVT-proteína y se incubaron durante otros 30 minutos. La mezcla se diluyó en tampón SPA sin o con RO7223280 frío 500 nM para medir la unión total y no específica respectivamente en una microplaca Optiplate-384 (Perkin Elmer). Cada pocillo contenía 25 ul de volumen total, proteína 18 nM, 5 % de dimetilsulfóxido y 6 % v/v de perlas de PVT. Las placas SPA se sellaron (TopSeal, Perkin Elmer) y se almacenaron a 4 °C durante la noche. Antes de la medición, las placas se mezclaron en un agitador durante 20 minutos, 750 rpm a temperatura ambiente y el sello se limpió minuciosamente con un spray antiestático para reducir los eventos electrostáticos. Los datos de centelleo se registraron con un Topcount NXT C384, en forma de tres réplicas independientes, cada una de las cuales consta de tres triplicados técnicos. La unión específica se calculó restando los recuentos brutos de unión no específica de los recuentos brutos de unión total. La constante de disociación Kd y la desviación estándar de las tres réplicas independientes se informan y se calcularon mediante la herramienta GraphPad Prism ‘Unión específica de un sitio’ .
Se llevaron a cabo experimentos de desplazamiento de radioligando de manera similar, aplicando una concentración constante de 25 nM [ 3H ]-RO7223280 y 16 concentraciones de ligandos fríos en presencia de LPS (Rc) 10 µM. Los valores de desplazamiento se normalizaron incluyendo nueve pocillos que no contenían radioligando (definido como 100 % de competencia) y nueve pocillos que contenían solo 8 uM de radioligando (definido como 0 % de competencia). La constante inhibidora Ki se calculó con la herramienta ‘One Site – Fit Ki’ utilizando la concentración (25 nM) y K d ( 86 nM) de [ 3H ]-RO7223280 como restricciones. El coeficiente de Hill se utilizó como métrica de control de calidad (teóricamente, n H = 1 para un inhibidor competitivo 1:1) y se determinó con la herramienta ‘[Inhibidor] vs. respuesta – Pendiente variable (cuatro parámetros)’.
Estamos frente a una interesante promesa de solución, que con mucha suerte podrá ver la luz comercial y la producción a escala el próximo año 2025, pero no sabemos a que precio, y esto es, no menor frente a una bacteria que puede matar a pacientes en nuestros hospitales y las dificultades financieras del sistema de salud, que por este tiempo, hasta definirse estas cuestiones terminar con la fase clínica, producción, precio, cobertura y accesibilidad, las medidas preventivas, la prevención de las neumonías de respirador, de la traslocación, del uso racional de antibióticos, nos obligará por dos años más a todos los que gestionamos responsabilizarnos por la salud de nuestros pacientes.
