Principal
En los últimos 30 años hemos presenciado avances asombrosos e incomparables en la investigación científica, desde una mejor comprensión de la fisiopatología de los procesos patológicos básicos y el desentrañar la maquinaria celular con resolución atómica hasta el desarrollo de terapias que alteran el curso y el resultado de las enfermedades en todas las áreas de la medicina. Además, los avances exponenciales en genómica, inmunología, proteómica, metabolómica, microbiomas intestinales, epigenética y virología en paralelo con la ciencia de big data, la biología computacional y la inteligencia artificial (IA) han impulsado estos avances. Además, el surgimiento de las tecnologías CRISPR–Cas9 ha abierto una tentadora variedad de oportunidades en la medicina personalizada.
A pesar de estos avances, su rápida traducción del laboratorio a la práctica clínica está retrasada en la mayoría de las áreas de la medicina y la investigación clínica sigue superada. El panorama del desarrollo de fármacos y los ensayos clínicos sigue siendo costoso para todas las partes interesadas, con una tasa de fracaso muy alta. En particular, la tasa de deserción de las terapias de desarrollo en etapa temprana es bastante alta, ya que más de dos tercios de los compuestos sucumben en el «valle de la muerte» entre el laboratorio y la práctica clínica 1 , 2 . Llevar un fármaco con éxito a través de todas las fases de desarrollo de fármacos hasta la clínica cuesta más de 1.500 a 2.500 millones de dólares (refs. 3 , 4 ). Esto, combinado con las ineficiencias y deficiencias inherentes que plagan el sistema de atención médica, está llevando a una crisis en la investigación clínica. Por lo tanto, se necesitan estrategias innovadoras para involucrar a los pacientes y generar la evidencia necesaria para impulsar nuevos avances en la clínica, de modo que puedan mejorar la salud pública. Para lograrlo, los modelos de investigación clínica tradicionales deben dar paso a ideas y diseños de ensayos de vanguardia.
Antes de la pandemia de COVID-19, la realización de investigaciones clínicas se había mantenido prácticamente sin cambios durante 30 años y algunas de las normas y reglas de realización de ensayos, aunque arcaicas, no se cuestionaban. La pandemia expuso muchas de las limitaciones sistémicas inherentes a la realización de ensayos 5 y obligó a la empresa de investigación de ensayos clínicos a reevaluar todos los procesos; por lo tanto, ha alterado, catalizado y acelerado la innovación en este ámbito 6 , 7 . Las lecciones aprendidas deberían ayudar a los investigadores a diseñar e implementar ensayos clínicos de próxima generación «centrados en el paciente».
Las enfermedades crónicas siguen afectando a millones de vidas y causando una gran presión financiera a la sociedad 8 , pero la investigación se ve obstaculizada por el hecho de que la mayoría de los datos residen en silos de datos. La subespecialización de la profesión clínica ha dado lugar a silos dentro y entre las especialidades; cada área de enfermedad importante parece funcionar de forma completamente independiente.
Sin embargo, la mejor atención clínica se proporciona de manera multidisciplinaria con toda la información relevante disponible y accesible. Una mejor investigación clínica debe aprovechar el conocimiento adquirido de cada una de las especialidades para lograr un modelo colaborativo que permita una atención multidisciplinaria de alta calidad y una innovación continua en medicina. Debido a que muchas disciplinas en medicina ven las mismas enfermedades de manera diferente (por ejemplo, los especialistas en enfermedades infecciosas ven la COVID-19 como una enfermedad viral, mientras que los expertos en cardiología la ven como una enfermedad inflamatoria), los enfoques interdisciplinarios deberán respetar los enfoques de otras disciplinas. Aunque un modelo único puede no ser apropiado para todas las enfermedades, la colaboración interdisciplinaria hará que el sistema sea más eficiente para generar la mejor evidencia.
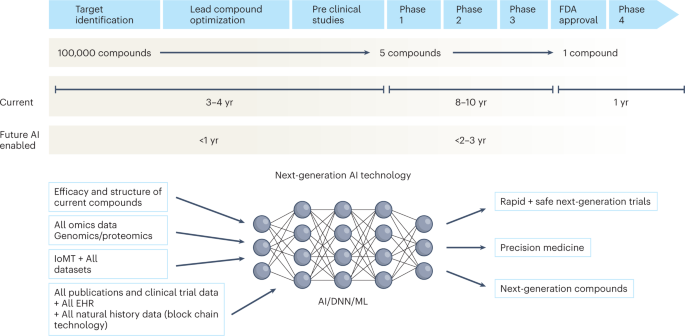
En la próxima década, la aplicación del aprendizaje automático, las redes neuronales profundas y la IA biomédica multimodal están preparadas para revitalizar la investigación clínica desde todos los ángulos, incluido el descubrimiento de fármacos, la interpretación de imágenes, la agilización de los registros médicos electrónicos, la mejora del flujo de trabajo y, con el tiempo, el avance de la salud pública (Fig. 1 ). Además, las innovaciones en wearables, tecnología de sensores y arquitecturas de Internet de las cosas médicas (IoMT) ofrecen muchas oportunidades (y desafíos) para adquirir datos 9 . En esta Perspectiva, comparto mi visión heurística del futuro de los ensayos clínicos y la generación de evidencia y delibero sobre las principales áreas que necesitan mejoras en los dominios del diseño de ensayos clínicos, la realización de ensayos clínicos y la generación de evidencia.
Diseño de ensayos clínicos
El diseño de los ensayos es uno de los pasos más importantes de la investigación clínica: un mejor diseño de los protocolos conduce a una mejor realización de los ensayos clínicos y a decisiones más rápidas de «seguir adelante o no». Además, las pérdidas derivadas de ensayos mal diseñados y fallidos no son solo financieras, sino también sociales.
Desafíos de los ensayos controlados aleatorios
Los ensayos controlados aleatorizados (ECA) han sido el estándar de oro para la generación de evidencia en todas las áreas de la medicina, ya que permiten estimaciones imparciales del efecto del tratamiento sin factores de confusión. Idealmente, cada tratamiento o intervención médica debería probarse mediante un ECA bien controlado y con potencia estadística. Sin embargo, la realización de ECA no siempre es factible debido a los desafíos para generar evidencia de manera oportuna, el costo, el diseño en poblaciones limitadas que impiden la generalización, las barreras éticas y el tiempo que lleva realizar estos ensayos. Cuando se completan y publican, los ECA se vuelven rápidamente obsoletos y, en algunos casos, irrelevantes para el contexto actual. Solo en el campo de la cardiología, 30.000 ECA no se han completado debido a los desafíos de reclutamiento 10 . Además, los ensayos se están diseñando de forma aislada y dentro de silos, y muchas preguntas clínicas quedan sin respuesta. Por lo tanto, los paradigmas de diseño de ensayos tradicionales deben adaptarse a los rápidos avances contemporáneos en genómica, inmunología y medicina de precisión 11 .
Avances en el diseño de ensayos clínicos
Se necesita evidencia de alta calidad para la práctica clínica, que tradicionalmente se ha logrado con ECA 12 . En la última década, se ha logrado un progreso sustancial en el diseño, la realización y la implementación de protocolos «maestros» (protocolos generales que se aplican a varios subestudios), lo que ha llevado a muchos cambios en la práctica que han mejorado sustancialmente el estancamiento de los ECA. Además, los protocolos maestros pueden implicar estudios intervencionistas paralelos en una sola enfermedad o múltiples enfermedades definidas por un biomarcador o entidad patológica 12 . Se incluyen cuatro clases diferentes de estudios en los protocolos maestros: el estudio paraguas, el estudio cesta, el estudio plataforma y el ensayo observacional maestro (MOT) (Fig. 2 ). Cada uno de estos es un diseño de ensayo único que puede incluir brazos independientes con intervenciones de control y puede analizarse individual y/o colectivamente, con mayor flexibilidad 13 , 14 . El campo de la oncología ha liderado estos esfuerzos más que cualquier otro campo, debido a los avances en genómica (para identificar alteraciones moleculares), el descubrimiento de terapias y la rápida traducción clínica, marcando así el comienzo de la era de la oncología de precisión.
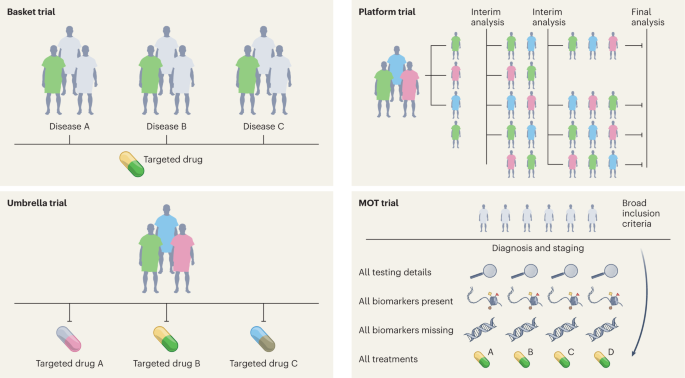
Estudio de paraguas
Los ensayos paraguas son diseños de estudio que evalúan múltiples terapias dirigidas para la misma entidad patológica, estratificadas por alteración molecular. Algunos ejemplos incluyen el ensayo de cáncer de mama I-SPY (Investigación de estudios seriados para predecir su respuesta terapéutica con imágenes y análisis molecular) y Lung-MAP (Protocolo maestro de cáncer de pulmón) 15 , 16 .
Prueba de canasta (o cubo)
Los ensayos de canasta son estudios independientes de la histología o agnósticos en los que se evalúa la terapia dirigida en múltiples tipos de enfermedades que albergan todas la misma aberración molecular subyacente. Por ejemplo, el estudio VE-Basket (en el que VE denota vemurafenib) 17 , el estudio Rare Oncology Agnostic Research (ROAR) 18 , el ensayo ARROW 19 y los ensayos LIBRETTO-001 20 , 21 han llevado a varias aprobaciones de medicamentos en poblaciones específicas impulsadas por biomarcadores de una manera dependiente e independiente de la histología.
Estudio de plataforma
Se trata de diseños de estudios multibrazo y multietapa que comparan varios grupos de intervención con un grupo de control común en el contexto del mismo protocolo maestro. Además, pueden ser diseños de estudios perpetuos/inmortales (sin fecha de finalización definida) y son más eficientes que los ensayos tradicionales debido al brazo de control compartido, que garantiza que una mayor proporción de pacientes se inscriban en los brazos intervencionistas/experimentales que en el brazo de control. El estudio de plataforma de evaluación aleatoria de la terapia de COVID-19 (RECOVERY) es un ejemplo destacado; este ensayo que cambió la práctica estableció la dexametasona como un tratamiento eficaz para COVID-19 (ref. 22 ) y también mostró que la hidroxicloroquina era ineficaz. Los estudios de plataforma son flexibles por diseño y no necesariamente necesitan tener un brazo de control compartido; la idea principal es que los brazos de intervención se pueden agregar a un ensayo en curso, por ejemplo, como en el ensayo de plataforma The UK Plasma Based Molecular Profiling of Advanced Breast Cancer to Inform Therapeutic CHoices (plasmaMATCH) 23 . Aunque los ensayos mencionados anteriormente se diseñaron en el contexto del desarrollo de fármacos en oncología y enfermedades infecciosas, el alcance de los ensayos de plataformas podría aprovecharse en otras áreas diversas, como la psicología clínica y la neurología 24 . Dichos ensayos también podrían utilizarse para intervenciones de salud mental digital y podrían implementarse fácilmente en entornos con recursos limitados 24 .
AGUDEZA
El MOT es un diseño de estudio observacional prospectivo que acepta ampliamente a los pacientes independientemente de la firma del biomarcador y recopila datos completos sobre cada participante 14 , 25 . El MOT es una combinación de los diseños de ensayo intervencionista maestro y ensayo observacional prospectivo e intenta hibridar el poder de los protocolos intervencionistas maestros basados en biomarcadores con la amplitud de los datos del mundo real (RWD) 14 , 25 . Este enfoque podría ser adecuado para recopilar RWD prospectivos en muchas especialidades; el Registro de resultados oncológicos asociados con pruebas y tratamiento (ROOT) MOT es un ejemplo 14 .
Desarrollo de biomarcadores y definición de puntos finales
El desarrollo de biomarcadores ha facilitado el progreso en el diseño de ensayos clínicos, con avances sin precedentes en genómica e inmunología que han dado lugar a varias aprobaciones de terapias dirigidas basadas en biomarcadores e inmunoterapia en la última década. De hecho, la evidencia genética humana brindó respaldo a más de dos tercios de las aprobaciones de medicamentos en 2021 (ref. 26 ). Los campos de la oncología y la genética se han beneficiado enormemente de estos avances, pero campos como la cardiología, la nefrología y la neumología aún están rezagados en las aprobaciones de medicamentos basados en biomarcadores.
Para acelerar el desarrollo de fármacos y los ensayos clínicos en todas las enfermedades importantes, tendremos que definir biomarcadores (ya sean clínicos, patológicos o fisiológicos) y su contexto de uso para cada proceso patológico y delinear puntos finales claros para los estudios 27 . Los biomarcadores pueden ser diagnósticos, pronósticos o predictivos y pueden informar el desarrollo temprano de fármacos, la selección de dosis y el diseño de ensayos. Además, los biomarcadores pueden ayudar a acelerar la ciencia básica y el descubrimiento de fármacos, todo ello con el objetivo final de mejorar la salud del paciente 28 . Sin embargo, el nivel de evidencia de un biomarcador depende en gran medida del contexto de uso.
Además de los biomarcadores, cada campo necesita definir áreas de máxima prioridad para la investigación e identificar los puntos finales más relevantes para responder a las preguntas de investigación prioritarias. Los puntos finales son medidas de salud y/o enfermedad y sirven para diferentes propósitos según la fase del ensayo 28 , 29 . Más allá de los puntos finales clínicos y regulatorios, los resultados informados por los pacientes y los puntos finales digitales también están surgiendo rápidamente.
Puntos finales digitales
Los puntos finales digitales son datos generados por sensores recopilados fuera del entorno clínico en el contexto de la vida cotidiana de los pacientes, como el uso de micrófonos de teléfonos inteligentes para monitorear el deterioro cognitivo en personas con enfermedad de Alzheimer o monitores de relojes inteligentes para evaluar el efecto de los medicamentos en personas con anemia de células falciformes 29 . Este es un área de considerable entusiasmo en medicina, ya que podría permitir un seguimiento más realista en el mundo real de la experiencia del paciente. Además, con el aumento de la realización de ensayos descentralizados en muchas especialidades, el monitoreo remoto está a punto de aumentar. Por ejemplo, un estudio reciente desarrolló un modelo de IA para detectar y rastrear la progresión de la enfermedad de Parkinson (para la que no hay biomarcadores) sobre la base de señales de respiración nocturna utilizando una evaluación no invasiva en el hogar, proporcionando evidencia de que la IA puede ser útil en la evaluación de riesgos antes del diagnóstico clínico de la afección 29 , 30 . Además, la detección digital de la fibrilación auricular mediante dispositivos inteligentes se ha evaluado ampliamente en estudios a gran escala, incluidos los estudios cardíacos de Apple 31 , Huawei 32 y Fitbit 33 . En total, estos estudios observacionales sin sedentarismo reclutaron a más de un millón de participantes, una hazaña asombrosa, y un estudio aleatorio mostró la superioridad de la detección digital de la fibrilación auricular sobre la atención habitual 34 .
La caracterización y evaluación digital del estado clínico debe estandarizarse y armonizarse, con la colaboración interdisciplinaria y el aporte de los reguladores. También se necesita consenso para identificar y caracterizar los puntos finales intermedios y sustitutos de las principales enfermedades crónicas. Esto requiere la incorporación específica de la especialidad de múltiples niveles de datos, como datos clínicos genómicos, proteómicos y basados en genotipo-fenotipo y mediciones específicas de la enfermedad, además de una capa de datos funcionales 26 . Los Institutos Nacionales de Salud (NIH) y la Administración de Alimentos y Medicamentos (FDA) han desarrollado recursos BEST (Biomarkers, EndpointS and other Tools) para aclarar la ambigüedad en los biomarcadores y los puntos finales. Este es un «documento vivo» que se actualiza continuamente a medida que cambian los estándares y la evidencia 35 y que aclara definiciones importantes y describe algunas de las relaciones jerárquicas, conexiones y dependencias entre los términos.
Realización de ensayos clínicos
Los componentes de la realización de ensayos clínicos son la implementación de protocolos; la selección, reclutamiento, monitoreo y retención de pacientes; la garantía del cumplimiento de los informes de seguridad; y la revisión y análisis continuos de datos. La industria farmacéutica y el sector de la atención médica invierten recursos sustanciales en la realización de ensayos clínicos, pero se necesitan cambios urgentemente para que el proceso sea más fluido. Además, el ritmo al que se realizan los ensayos clínicos es demasiado lento para seguir los avances de la investigación en todos los campos; por lo tanto, se necesita una transformación de alta tecnología de cada componente de manera gradual.
Uno de los aspectos positivos de la pandemia es que obligó al sistema a reorientar los ensayos clínicos para que estuvieran más centrados en el paciente que antes, dando así más importancia al principal sujeto de la investigación clínica: el paciente 36 (Fig. 3 ). Esto ha dado lugar a ensayos descentralizados y ensayos digitales, remotos y «virtuales» (que permiten a los pacientes acceder a los ensayos independientemente de su ubicación geográfica), así como a conceptos de «hospital en casa» y de seguimiento en el hogar 37 . Estos rápidos avances se han visto facilitados por la orientación de las autoridades reguladoras 38 . La adopción de un enfoque basado en la IA para mejorar la experiencia del paciente puede mejorar aún más las evaluaciones de alta fidelidad y garantizar el cumplimiento de los protocolos 39 . Aunque la digitalización, la virtualización y la descentralización no son curas para las crisis de la investigación clínica, pueden crear eficiencias que pueden tener un impacto considerable y a largo plazo.
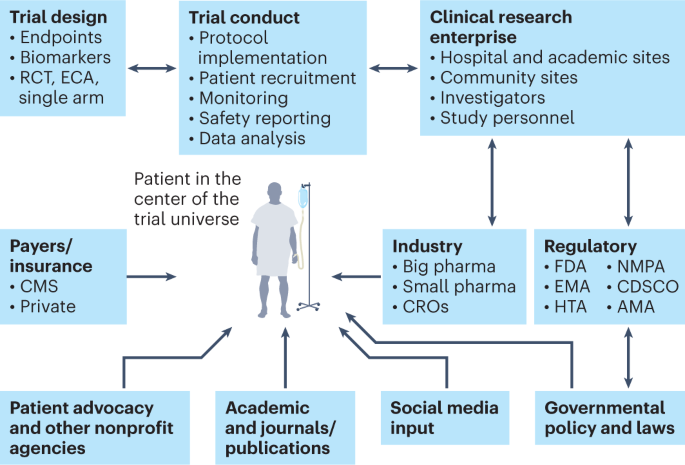
Los médicos, los miembros del equipo de atención médica y los investigadores clínicos de los centros académicos y otros sitios de inscripción de pacientes contribuyen enormemente al reclutamiento de pacientes. Además, los sitios de alto impacto y alto funcionamiento (como los principales centros académicos de excelencia) a menudo tienen una cartera de ensayos y examinan a los pacientes que se presentan al sistema de manera eficiente. Sin embargo, estos sitios son una minoría y la mayoría de los sitios de ensayos clínicos enfrentan limitaciones de personal y otras barreras.
Empresa de investigación de ensayos clínicos
La eficiencia y la colaboración en la empresa de investigación de ensayos clínicos son componentes importantes para el éxito de los ensayos clínicos. Los principales componentes de la empresa de ensayos clínicos son los pacientes, los centros académicos, los patrocinadores de la industria (grandes y pequeñas empresas farmacéuticas), los patrocinadores gubernamentales/grupos cooperativos, las agencias reguladoras, las organizaciones de defensa de los pacientes y las organizaciones de investigación por contrato (CRO), y todos ellos deben trabajar juntos con el paciente como el centro del universo de los ensayos clínicos (Fig. 3 ). Además, todo este sistema necesita una revisión digital, ya que muchos sitios todavía utilizan carpetas de protocolos, diarios con lápiz y papel, faxes entre sitios, datos no estructurados y sistemas de software con décadas de antigüedad. Los ensayos clínicos de registro deben gestionarse bien día a día con una rigurosa captura y seguimiento electrónico de datos. La integración de la tecnología blockchain en el sistema de gestión de ensayos clínicos podría reforzar la confianza en el proceso de ensayos clínicos y facilitar la supervisión regulatoria 40 .
La participación de los pacientes en los ensayos clínicos es fundamental, ya que no puede haber ensayos sin pacientes. Los organizadores de los ensayos clínicos deben facilitar la participación de los pacientes en los ensayos. Además, las decisiones de tratamiento médico-paciente para las enfermedades importantes deben incluir opciones de ensayos clínicos como estándar. Estos ensayos clínicos deben ser de fácil acceso y deben garantizar que ningún paciente sea excluido innecesariamente; esto se puede lograr con servicios de búsqueda y emparejamiento de ensayos clínicos independientes del sitio. Además, la capacitación en ensayos clínicos debe ser parte de la educación médica para que un grupo diverso de investigadores y personal capacitado de toda la empresa de atención médica pueda estar disponible para la investigación clínica.
Ya era hora
Los plazos de desarrollo clínico de los fármacos candidatos son una carrera contra el tiempo desde que se presentan las patentes hasta la aprobación final de la FDA 41 . Los plazos de desarrollo de fármacos, en promedio, son de aproximadamente 10 años (Fig. 1 ). La rapidez del desarrollo de las vacunas de ARNm de COVID-19 y el tratamiento oral COVID-19 en tabletas de nirmatrelvir/ritonavir, ambos desarrollados en un año utilizando un «enfoque de velocidad de la luz», no debería ser una excepción 42 . Las lecciones aprendidas deberían proporcionar un modelo para múltiples áreas terapéuticas de necesidades no satisfechas. Las dos pequeñas moléculas que tienen el récord del plazo más corto en el desarrollo de fármacos, osimertinib para el cáncer de pulmón de células no pequeñas (CPCNP) con mutación de EGFR (984 días mediante aprobación acelerada) y elexacaftor para la fibrosis quística (1.043 días mediante la vía regular) 41 , en tiempos no pandémicos demuestran que esto es posible.
Los obstáculos regulatorios que ralentizan el desarrollo de medicamentos hicieron necesaria la creación de programas como el programa de aprobación acelerada de la FDA, que se introdujo en 1992 para abordar la crisis del VIH y el SIDA y que desde entonces ha beneficiado a áreas altamente especializadas como la oncología de precisión 43 . Se han creado múltiples programas para acortar los plazos del proceso previo a la comercialización, incluidos los de revisión prioritaria, designación de vía rápida, designación de medicamento innovador y designación de medicamento huérfano 44 . Sin embargo, más allá de estos programas, los plazos siguen siendo lentos y existe una necesidad urgente de abordar este problema para todas las enfermedades, ya que la velocidad del desarrollo de medicamentos es crucial tanto para los pacientes como para los médicos y las partes interesadas en el desarrollo de medicamentos.
Globalización del desarrollo, la armonización y la transportabilidad de medicamentos
Aunque el mandato de la FDA se dirige a la población estadounidense, su influencia es global y, funcionalmente, la FDA es el regulador de facto para el mundo. Otras autoridades reguladoras como la Agencia Europea de Medicamentos, la Administración Nacional de Productos Médicos en China y la Organización Central de Control de Estándares de Medicamentos en la India, que en total atienden a más de 3 mil millones de la población mundial, también están evolucionando como actores clave en el sector farmacéutico global. Además, la recién establecida Agencia Africana de Medicamentos se creó (en 2019) para acelerar los plazos de aprobación de vacunas y medicamentos y mejorar el acceso a los medicamentos, especialmente para enfermedades infecciosas emergentes endémicas del continente 45 . Todas estas agencias deben poder valerse por sí solas. Además, existe una necesidad urgente de armonización global entre las autoridades reguladoras para abordar las desigualdades sustanciales en el acceso a los medicamentos. Idealmente, los ensayos clínicos para nuevas terapias deberían realizarse a nivel mundial, para el acceso y la generalización 46 . Sin embargo, la realidad es que los ensayos clínicos, incluidos los ECA, no se pueden realizar en todos los países para generar evidencia específica para la población de ese país. La generación de evidencia mediante el análisis de transportabilidad está ganando terreno y se refiere a la capacidad de generalizar inferencias de una muestra de estudio en un país a una población objetivo en otro país donde no se realizó el estudio 47 , 48. Los análisis de transportabilidad pueden ofrecer cierta evidencia de validez externa con implicaciones para las evaluaciones regulatorias y de tecnología sanitaria locales 48 .
Generación de evidencia en la investigación clínica
Estudios clínicos de enfermedades raras
A medida que los avances científicos impulsan los ensayos clínicos, se están diseñando y llevando a cabo ensayos sobre cánceres y muchas enfermedades raras en pequeños subconjuntos definidos genéticamente o definidos por biomarcadores. Además, los nuevos métodos para generar evidencia de beneficio clínico pueden acelerar la realización de ensayos clínicos y proporcionar a las personas con enfermedades raras acceso a nuevos compuestos terapéuticos. Se estima que las enfermedades raras afectan a un estimado de 263 millones a 446 millones de personas en todo el mundo en un momento dado y se están convirtiendo cada vez más en una enorme carga de salud pública 49 . Los ensayos clínicos en este contexto vienen con sus propios desafíos derivados de la rareza de las condiciones y los datos incompletos de la historia natural 50 . Sin embargo, los avances notables en biología molecular junto con la legislación para estimular el desarrollo de terapias para enfermedades huérfanas han llevado al progreso. Existe una creciente flexibilidad regulatoria para utilizar programas como el programa de aprobación acelerada, y hay casos en los que los ensayos han utilizado brazos de control externos basados en RWD.
Como ejemplo, la FDA otorgó la aprobación acelerada a alpelisib (Vijoice, Novartis) para adultos y niños mayores de 2 años que requieren terapia sistémica para el espectro de sobrecrecimiento relacionado con PIK3CA, que incluye un grupo de trastornos raros vinculados a mutaciones en el gen PIK3CA 51 . Curiosamente, la eficacia se evaluó utilizando una revisión retrospectiva de historias clínicas de RWD de EPIK-P1 ( NCT04285723 ), un estudio clínico de un solo brazo en el que individuos con espectro de sobrecrecimiento relacionado con PIK3CA recibieron alpelisib como parte de un programa de acceso ampliado para uso compasivo. La solicitud para esta aprobación utilizó el programa piloto de Revisión Oncológica en Tiempo Real 52 , que agilizó el envío de datos antes de presentar la solicitud clínica completa, y la Ayuda de Evaluación 53 , una presentación voluntaria del solicitante para facilitar la evaluación por parte de la FDA. Como resultado, a esta solicitud se le otorgó revisión prioritaria, designación innovadora y designación de medicamento huérfano 51 .
Ensayos N de 1
En la era de la medicina genómica individualizada, los ensayos N-of-1 están surgiendo como una herramienta para estudiar enfermedades raras potencialmente fatales. El ensayo N-of-1 es un ensayo clínico de un solo paciente que utiliza a la persona individual como una unidad de investigación para evaluar la eficacia y/o los eventos adversos de diferentes intervenciones a través de criterios objetivos basados en datos 54 . Por ejemplo, se diseñó y evaluó una terapia con oligonucleótidos antisentido para un solo paciente que tenía un trastorno neurodegenerativo genético fatal conocido como lipofuscinosis ceroidea neuronal CLN7 (una forma de la enfermedad de Batten) 55 . Otro paciente (que resultó ser médico) con enfermedad de Castleman idiopática refractaria a la terapia con bloqueo de IL-6 identificó la alteración molecular causal en su propia enfermedad para desarrollar una terapia personalizada 56 . En otro ejemplo, se evaluó la escalada rápida de dosis con un inhibidor selectivo de RET en un solo paciente con carcinoma medular de tiroides altamente refractario, para superar un mecanismo de resistencia específico de ese paciente 57 .
Estos nuevos y sensacionales paradigmas de descubrimiento y traducción de fármacos plantean preguntas importantes, como qué nivel de evidencia se necesita antes de exponer a un ser humano a un nuevo fármaco, qué evidencia podría generar este enfoque para el próximo paciente y qué desafíos podrían existir con la generalización 58 . El concepto de «gemelos digitales» específicos del paciente análogos médicos es un área emergente de investigación que tiene el potencial de combinar datos polinómicos (datos mecanicistas, historial médico, con el poder de la IA) y tal vez sirva para mejorar los ensayos N-de-1 en el futuro, para personalizar aún más la medicina 37 , 59 , 60 .
RWD y evidencia del mundo real
Una de las principales críticas a toda la investigación de ensayos clínicos es que los ensayos clínicos no representan a la población del «mundo real»; a menudo, los criterios restrictivos de los ensayos clínicos y los análisis limitados enmarcados para responder preguntas específicas pueden no ser aplicables a los pacientes del mundo real. Por lo tanto, existe una gran brecha entre el mundo de los ensayos y el mundo real, y se han hecho intentos para cerrar esta brecha 61 . Los ensayos convencionales se han diseñado sobre la base de la idea errónea de que los organismos reguladores pueden no dar cabida a evidencia más moderna y diversa de los datos de los ensayos clínicos, lo que ya no es el caso 61 , 62 .
Es importante distinguir entre RWD, que se refiere a los datos generados a partir de la atención rutinaria y estándar de los pacientes 62 , y la «evidencia» del mundo real (RWE), que es la evidencia generada a partir de RWD con respecto al uso potencial de un producto. La RWE se genera mediante diseños de ensayos o análisis y no se limita a ensayos aleatorios; en cambio, proviene de ensayos pragmáticos y estudios observacionales prospectivos y/o retrospectivos 62 , 63 .
En este ámbito de los datos de salud y medicinas, todas las partes interesadas buscan la orientación de los reguladores. En consecuencia, los reguladores han adoptado un enfoque práctico y han proporcionado orientación y un marco integral lanzado a través de la Ley de Curas del Siglo XXI 62 , 64 . Además, la FDA utiliza los datos de salud y medicinas para el monitoreo de seguridad posterior a la comercialización, y las agencias de seguros han comenzado a utilizar dichos datos para las decisiones de cobertura 62 . Esto ha sido necesario debido a la rápida aceleración de la entrada de datos de múltiples flujos y capas en los registros médicos electrónicos, así como en los dispositivos portátiles y los biosensores, en paralelo con nuevas capacidades analíticas (IA multimodal) para analizar la enorme cantidad de datos.
Evidencia de brazos de control sintéticos o externos
Los RCT se consideran el estándar de oro para el desarrollo de fármacos y la evidencia, ya que permiten la estimación de los efectos del tratamiento que se pueden asignar al brazo experimental de interés. La aleatorización en estos estudios reduce la preocupación por el sesgo de confusión al eliminar los desequilibrios sistemáticos entre los brazos en los factores pronósticos medidos y no medidos 65 . Sin embargo, los avances en la genómica de las enfermedades raras y el descubrimiento de cánceres raros impulsados por oncogenes han llevado a terapias dirigidas específicas, para las cuales la evaluación en RCT puede no ser factible o ética y puede retrasar el acceso de los pacientes a terapias prometedoras o que salvan vidas.
En tales casos, los brazos de control sintéticos están surgiendo como opciones para generar brazos de comparación que puedan «imitar» los brazos de comparación de los RCT. Los brazos de control sintéticos son externos al estudio en cuestión, y la mayoría se derivan de RWD 65 . Además, los RWD se obtienen de registros médicos electrónicos, datos de reclamaciones administrativas, registros de historia natural y datos generados por pacientes de muchas fuentes, incluidos los dispositivos portátiles 65 . Los brazos de control sintéticos también pueden generarse a partir de datos de ensayos clínicos anteriores (ensayos individuales o agrupados). Esta es un área emergente preparada para la innovación, ya que ahora hay muchos datos disponibles de múltiples fuentes.
El CPCNP se divide cada vez más en pequeños subconjuntos impulsados por oncogenes, lo que dificulta la realización de ensayos aleatorizados 66 , y los desarrollos recientes en el panorama de los ensayos de CPCNP ilustran la utilidad de los brazos de control sintéticos. Por ejemplo, las fusiones de RET son impulsores genómicos en el 1-2% de los CPCNP, y el pralsetinib es una terapia selectiva dirigida a RET que muestra respuestas prometedoras incluso en individuos con enfermedad avanzada. El estudio ARROW ( NCT03037385 ) fue un ensayo de registro de un solo brazo, realizado a nivel mundial, para evaluar el pralsetinib en individuos con fusión de RET positiva con CPCNP 67 , 68 . Este ensayo mostró un beneficio relativo de supervivencia con el fármaco en comparación con un brazo de control externo de estándar de atención que consistía en cohortes de RWD derivadas de dos bases de datos de Flatiron Health 66 . Una plantilla para futuros estudios de esta naturaleza que utilicen análisis de sesgo cuantitativo mostró que las comparaciones entre el grupo de control externo y el grupo de prueba son sólidas y capaces de soportar problemas como la falta de datos, resultados potencialmente peores en RWD y confusión residual 66 . En general, el estudio proporcionó evidencia a favor de pralsetinib como tratamiento de primera línea para el CPNM con fusión RET positiva.
El uso de brazos de control sintéticos puede acelerar el desarrollo de fármacos, y el escepticismo inicial sobre ellos surgió principalmente de una falta de precedentes y dirección de las autoridades regulatorias. Estas preocupaciones ahora se están disipando a medida que los brazos de control sintéticos se han utilizado recientemente para aprobaciones de fármacos para enfermedades ultra raras. Por ejemplo, la neurofibromatosis es una enfermedad rara que se observa en 1 de cada 3000 nacimientos. Los pacientes desarrollan lesiones de neurofibroma plexiforme que son dolorosas y debilitantes, y causan disfunción motora y neuronal. El inhibidor de MEK selumetinib fue aprobado para pacientes pediátricos con neurofibromas plexiformes sintomáticos e inoperables sobre la base de un conjunto de datos de 50 pacientes del Selumetinib en el ensayo de neurofibroma pediátrico (SPRINT), un ensayo de fase 2 de un solo brazo que muestra una tasa de respuesta objetiva duradera y mejoras en los síntomas funcionales 65 , 69 , 70 . Los brazos de comparación de dos ensayos realizados previamente proporcionaron evidencia de la historia natural de la enfermedad y se presentaron como un brazo de control externo, lo que ayudó a confirmar que las regresiones espontáneas eran poco comunes y que las respuestas observadas y la mejoría de los síntomas representaban un efecto genuino del tratamiento 69 .
A pesar de este progreso, los brazos de control externo siguen siendo un concepto emergente y se han utilizado principalmente para investigar la historia natural de la enfermedad y, por lo general, no se han incluido como evidencia primaria o en las etiquetas de los productos. Sin embargo, en el futuro, puedo imaginar este análisis de efectividad comparativa y los brazos de comparación como evidencia primaria para respaldar la aprobación de medicamentos. Los desafíos surgen principalmente de la calidad de los datos y la falta de datos, así como de la incertidumbre sobre si los datos de control externo son adecuados para el propósito. Sin embargo, algunas de estas preocupaciones se pueden mitigar mediante el análisis de sesgo cuantitativo y otras metodologías 66 , 71 .
Ensayos clínicos pediátricos
Aunque la investigación pediátrica ha estado a la vanguardia de los principales avances en medicina (la oxigenación por membrana extracorpórea 72 es un ejemplo notable) y ha ampliado los límites de la oncología moderna (por ejemplo, en el tratamiento de la leucemia pediátrica), las innovaciones en el desarrollo de nuevos fármacos a menudo se retrasan. Muchas enfermedades raras y huérfanas ocurren principalmente en la población pediátrica, y el desarrollo de fármacos en esta población siempre ha sido un desafío operativo, ético, estadístico y metodológico 73 , 74 . Esto se ve agravado por la comprensión limitada de la biología básica, la ontología de las manifestaciones de la enfermedad y la seguridad aguda y a largo plazo de los productos 73 , 74 . Además, existe un uso considerable fuera de etiqueta de productos en niños muy pequeños, lactantes y neonatos donde los ensayos clínicos no han sido factibles, y es imperativo que se genere evidencia de alto nivel mediante métodos creativos. Programas como la Ley de Mejores Medicamentos para Niños (en 2002) y la Ley de Equidad en la Investigación Pediátrica (en 2003), que se hicieron permanentes en 2012 bajo la Ley de Seguridad e Innovación de la FDA, han incentivado y mejorado el desarrollo de terapias pediátricas 73 . Los diseños de ensayos innovadores, los datos de investigación y el aprovechamiento de datos de otros recursos pueden ayudar con la evaluación de riesgos y beneficios y la aprobación de medicamentos, como la aprobación para la neurofibromatosis tipo 1 (NF1) 73 .
Reimaginando el futuro de los ensayos clínicos
AI
El panorama de la IA en medicina se ha transformado recientemente y está a punto de volverse omnipresente. Varios RCT han cuantificado los beneficios de la IA en especialidades que utilizan el reconocimiento de patrones y la interpretación de imágenes, como radiología (mamografía y detección de cáncer de pulmón), cardiología (interpretación de electrocardiogramas (ECG), evaluación funcional cardíaca y detección de fibrilación auricular), gastroenterología (interpretación de colonoscopias), patología (diagnóstico de cáncer), neurología (seguimiento de la evolución de la esclerosis lateral amiotrófica y la enfermedad de Parkinson), dermatología (diagnóstico de lesiones) y oftalmología (detección de enfermedades oculares) 75 . Sin embargo, la mayoría de la investigación de IA se centra en aplicaciones de «prestación de atención clínica» y no en «investigación de ensayos clínicos» 76 .
La integración de la IA en la investigación de ensayos clínicos ha sido más lenta de lo esperado, debido principalmente a la fricción (percibida) entre la IA y la inteligencia humana. Sin embargo, se deben realizar ensayos de generación e interpretación de datos, y la IA se debe utilizar para aumentar la inteligencia humana, no como algo que la reemplace 77 . Los ensayos clínicos de próxima generación que utilicen IA deben considerar escenarios de IA + humanos en lugar de IA versus humanos 75 , 78 . Las pautas de ensayos clínicos para protocolos (extensión de Elementos de protocolo estándar: Recomendaciones para ensayos intervencionistas – Inteligencia artificial (SPIRIT-AI)) y publicaciones (extensión de Estándares consolidados para la presentación de informes de ensayos – Inteligencia artificial (CONSORT-AI)) 79 , 80 tienen como objetivo lograr informes estandarizados y transparentes para ensayos clínicos aleatorizados que involucran IA, y estos son solo el comienzo de una nueva fase de modernización de la investigación clínica.
Teniendo en cuenta el tiempo y el coste que supone desarrollar un fármaco, cada fármaco que fracasa en el mercado representa una pérdida considerable para el ecosistema de desarrollo de fármacos. Además, los diseños de ensayos de mala calidad, el reclutamiento de pacientes subóptimo, la infraestructura deficiente para realizar ensayos y la ineficiencia en la realización y el seguimiento de los ensayos han plagado el sistema durante décadas. La IA tiene el potencial de mejorar todas las fases del desarrollo de fármacos, desde el diseño del fármaco hasta el ciclo completo de desarrollo del fármaco (Fig. 1 ).
La realización de ensayos clínicos todavía es rudimentaria en muchos sentidos. Por ejemplo, en los ensayos oncológicos, se miden y se siguen a lo largo del tiempo algunos aspectos de las lesiones bidimensionales y se evalúa la eficacia del fármaco mediante la reducción de estas lesiones. Las evaluaciones cuantitativas automatizadas y las redes neuronales artificiales pueden ayudar en el procesamiento rápido y automatizado de múltiples lesiones 81 . En los ensayos de cardiología, los signos vitales se miden una vez a la semana en la clínica y, en neurología, se administran cuestionarios a los pacientes en la clínica. Ahora, todos estos datos se pueden rastrear de forma dinámica en tiempo real utilizando tecnología de sensores portátiles. La aplicación de IA a dichas áreas puede tener un impacto transformador a corto plazo. Además, el reconocimiento de patrones mediante redes neuronales profundas puede ayudar con la lectura de exploraciones, imágenes patológicas y electrocardiogramas, entre otros 37 , 78 .
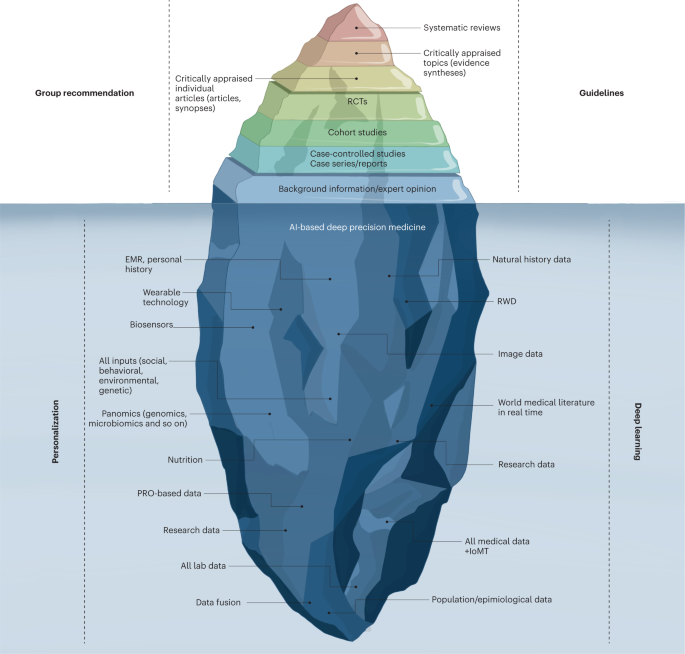
La pirámide actual de la medicina basada en la evidencia representa la punta del iceberg y apenas proporciona evidencia superficial para atender a un paciente genérico (Fig. 4 ). Por lo tanto, se necesita una síntesis y amalgama profunda de todos los datos disponibles para lograr una medicina basada en la evidencia «profunda» de próxima generación. El principal desafío en las próximas dos décadas será aprovechar el potencial de la generación de evidencia multidimensional 82 mediante la extracción, cotejo y minería de grandes conjuntos de datos de historia natural, genómica y todos los demás análisis ómicos, todos los estudios clínicos publicados, RWD, datos de dispositivos inteligentes ubicuos y datos acumulados del IoMT para proporcionar evidencia de próxima generación para la medicina profunda.
Alianzas para el desarrollo de medicamentos
En la actualidad, la industria farmacéutica es el principal impulsor del desarrollo de fármacos, y sus gastos superan con creces las inversiones de cualquier agencia nacional como los Institutos Nacionales de Salud 61 . Hay dos dominios de ensayos clínicos. El primero de ellos es el de las «grandes farmacéuticas», que utilizan CRO para realizar ensayos; dichos ensayos son muy a menudo aprobados para registro por la FDA. El segundo dominio abarca los ensayos clínicos académicos, que a menudo operan con un presupuesto muy limitado, no suelen evaluar nuevos compuestos y, por lo tanto, rara vez dan lugar al registro de la FDA. En esta era de reducción de la financiación federal para la investigación, se necesitan más asociaciones para el desarrollo de fármacos. Los centros académicos y los sitios comunitarios son cruciales para la inscripción de pacientes; sin embargo, una mentalidad compartimentada ha afectado al desarrollo de fármacos y ha retrasado el acceso a terapias que salvan vidas. Por lo tanto, las colaboraciones entre organizaciones de enfermedades específicas, instituciones académicas, agencias federales y grupos de defensa de los pacientes son cruciales para mejorar la salud de las poblaciones (Fig. 3 ). Debido a que la industria farmacéutica duda en invertir grandes cantidades con un rendimiento financiero limitado, especialmente en enfermedades raras, las agencias federales han desarrollado programas para incentivar el desarrollo de medicamentos para enfermedades raras 1 . Además, las organizaciones centradas en enfermedades han colaborado con la industria farmacéutica, las agencias federales y la academia para formar «filantropía de riesgo» con modelos financieros de reparto de riesgos para reducir el riesgo del desarrollo de medicamentos 1 . Muchas instituciones académicas están entrando en alianzas estratégicas de reparto de riesgos con la industria farmacéutica para colaborar en las fases de desarrollo preclínico y clínico. Estos modelos de asociación innovadores y exitosos han sentado un precedente en enfermedades como la fibrosis quística, el mieloma múltiple, la diabetes mellitus tipo 1 y otras enfermedades raras 1 . Estas colaboraciones han catalizado eficazmente la innovación a través de todas las fases del desarrollo de medicamentos y han proporcionado una razón convincente para sostener y fomentar más programas de este tipo.
Investigación sobre redes sociales y comunidades en línea
Las redes sociales (Twitter, Facebook, etc.) pueden influir en la incorporación de pacientes a los ensayos clínicos. Pueden influir en gran medida y abordar los desafíos históricos de los ensayos clínicos, incluida la falta de conocimiento entre los pacientes y los médicos sobre los ensayos disponibles y la falta de participación de la comunidad. Más de 4.480 millones de personas utilizan las redes sociales en todo el mundo, y se prevé que esta cifra aumente a casi 6.000 millones en 2027 (ref. 83 ). Más del 70% de los estadounidenses están en las redes sociales, incluidos los habitantes rurales y las poblaciones de adolescentes y adultos jóvenes que siempre han estado subrepresentados en los ensayos clínicos. Aunque muchos adultos mayores no utilizan las redes sociales, es probable que sus cuidadores sí lo hagan.
Las personas con enfermedades terminales suelen experimentar por sí mismas con medicamentos, y las comunidades de pacientes en línea pueden proporcionar entornos para compartir y monitorear el uso de dichos medicamentos. Esto puede permitir que se planifiquen estudios observacionales en torno a datos de resultados cuantitativos basados en Internet. Por ejemplo, los investigadores desarrollaron un algoritmo para diseccionar los datos informados en el sitio web PatientsLikeMe por personas con esclerosis lateral amiotrófica que experimentaron con el tratamiento con carbonato de litio 84 . Este análisis llegó a la misma conclusión que un RCT posterior, lo que sugiere que los datos del comportamiento de los pacientes en línea pueden ayudar a acelerar el desarrollo de medicamentos y evaluar la efectividad de los medicamentos que ya se utilizan.
Un aumento de la participación de los pacientes y de los grupos de apoyo a los pacientes puede ayudar a la educación y la divulgación de los pacientes y puede facilitar la investigación en colaboración con los pacientes, además de permitir la incorporación de las perspectivas de los pacientes en el diseño de la investigación clínica, generando en última instancia una investigación impulsada por las necesidades de personas reales con la enfermedad en investigación. Además, las redes sociales rompen los silos que dividen a los investigadores y los médicos, creando un enorme potencial para influir en todas las áreas de la medicina 85 .
Conclusión
El éxito de los ensayos clínicos futuros requiere una transformación fundamental en la forma en que se diseñan, realizan, monitorean, adaptan, informan y regulan los ensayos para generar la mejor evidencia. El modelo del statu quo es insostenible. En cambio, se necesita una medicina preventiva, personalizada, pragmática y participativa del paciente, y se requieren cambios de paradigma para llegar a ella mediante un crecimiento sostenible. Es necesario romper los silos. Los estándares de atención y los ensayos clínicos se consideran actualmente en ámbitos diferentes; sin embargo, el objetivo general de ambos es mejorar los resultados de salud. La pandemia de COVID-19 creó una oportunidad para observar cómo la atención clínica de rutina y los ensayos clínicos pueden trabajar sinérgicamente para generar evidencia 86 . Los ensayos de plataforma pragmática como el ensayo RECOVERY deben ser un modelo y una guía para la eficiencia de los ensayos y el impacto en tiempo real.
Los paradigmas actuales deben ser desafiados continuamente por la tecnología emergente y por todas las partes interesadas (las nuevas generaciones de científicos, médicos, la industria farmacéutica, las autoridades regulatorias y, lo más importante, los pacientes). La innovación disruptiva debe llevar a que cada sitio clínico sea un sitio de investigación, con todos los controles de calidad necesarios y la investigación como parte del estándar de atención. El sistema de atención médica debe integrarse en un sistema intuitivo de generación de RWE, con la investigación clínica y la atención clínica yendo de la mano. Más allá de un destello creativo ad hoc (necesario por una pandemia), se necesitará un impulso sostenido para aprovechar el conocimiento adquirido de programas como «Operación Warp Speed» (iniciada por el gobierno de EE. UU. para acelerar el desarrollo de la vacuna COVID-19). Mi opinión personal es que cada enfermedad importante necesita un programa «Moonshot» y cada enfermedad rara debería tener una «Operación Warp Speed», ambos con objetivos claramente identificados y sostenibles para mejorar la salud de la población y abordar la equidad, la diversidad y el acceso global a las terapias. Los avances metodológicos y los futuros análisis de todos los datos basados en inteligencia artificial proporcionarán evidencia profunda para lograr el objetivo de la medicina personalizada, es decir, ofrecer el tratamiento adecuado al paciente adecuado en el momento adecuado.
