- Manik Garg , Marcin Karpinski ,Dorota Matelska , Lawrence Middleton , Oliver S. Burren , Fengyuan Hu , Eleanor Wheeler ,Katherine R. Smith ,Margarete A. Fabre ,Jonathan Mitchell ,Amanda O’Neill ,Euan A. Ashley ,Andrew R. Harper ,Rey Quanli , Ryan S. Dhindsa ,
La aparición de conjuntos de datos a nivel de biobanco ofrece nuevas oportunidades para descubrir nuevos biomarcadores y desarrollar algoritmos predictivos para enfermedades humanas. Aquí, presentamos un marco de aprendizaje automático en conjunto (aprendizaje automático con asociaciones de fenotipos, MILTON) que utiliza una variedad de biomarcadores para predecir 3213 enfermedades en el Biobanco del Reino Unido. Aprovechando los datos de registros de salud longitudinales del Biobanco del Reino Unido, MILTON predice casos de enfermedades incidentes no diagnosticados en el momento del reclutamiento, superando ampliamente las puntuaciones de riesgo poligénico disponibles. Demostramos además la utilidad de MILTON para aumentar los análisis de asociación genética en un estudio de asociación de todo el fenoma de 484.230 muestras secuenciadas del genoma, junto con 46.327 muestras con datos de proteómica plasmática coincidentes. Esto dio como resultado señales mejoradas para 88 relaciones gen-enfermedad conocidas ( P < 1 × 10 −8 ) junto con 182 relaciones gen-enfermedad que no alcanzaron significación a nivel de todo el genoma en las cohortes de referencia no aumentadas. Validamos estos descubrimientos en el biobanco FinnGen junto con dos métodos de aprendizaje automático ortogonales diseñados para la priorización de genes y enfermedades. Todas las asociaciones entre genes y enfermedades extraídas y los biomarcadores predictivos de enfermedades incidentes están disponibles públicamente
Identificar a las personas con alto riesgo de desarrollar enfermedades es una prioridad para la medicina preventiva. La mayoría de las herramientas tradicionales de evaluación de riesgos se basan en parámetros clínicos como la edad, el sexo y los antecedentes familiares, y en un conjunto reducido de biomarcadores básicos adaptados a la enfermedad en estudio 1 , 2 , 3 . Sin embargo, estas herramientas pueden no captar el espectro completo de procesos biológicos que subyacen a enfermedades complejas. La aparición de biobancos a gran escala que integran registros médicos electrónicos y datos multiómicos (como análisis de sangre estándar, proteómica y metabolómica) brindan una oportunidad sin precedentes para descubrir nuevos biomarcadores y colecciones de biomarcadores para predecir mejor la aparición de enfermedades.
El Biobanco del Reino Unido (UK Biobank, UKB; https://www.ukbiobank.ac.uk ) es una de las cohortes de biobancos más grandes, que incluye datos de historiales médicos y secuenciación genética de medio millón de personas de entre 40 y 69 años en el momento del reclutamiento. Hay un amplio catálogo de datos fenotípicos para cada participante, que incluye datos de historiales médicos actualizados continuamente, mediciones biométricas, indicadores de estilo de vida, biomarcadores de sangre y orina e imágenes. Este biobanco ha revelado una variedad de nuevas asociaciones genéticas y candidatos a objetivos terapéuticos 4 , 5 , 6 . Debido a su escala, el UKB también ofrece la oportunidad de identificar combinaciones de biomarcadores que pueden predecir mejor la aparición de la enfermedad que cualquier biomarcador solo. Por ejemplo, un artículo reciente utilizó ~1500 mediciones de proteínas plasmáticas de más de 50 000 participantes del UKB para identificar un puñado de proteínas que podrían predecir con precisión la demencia incluso 10 años antes del diagnóstico 7 . Esto sugiere que un enfoque sistemático en todos los fenotipos bien representados en el UKB podría descubrir muchos otros modelos precisos de predicción del riesgo de enfermedades.
Más allá de la predicción de enfermedades y el descubrimiento de biomarcadores, las predicciones basadas en biomarcadores de individuos con enfermedades también podrían aumentar los análisis de descubrimiento genético de casos y controles. Más específicamente, una limitación de la mayoría de los biobancos es que las definiciones de fenotipo a menudo se basan en códigos de facturación y autoinformes, lo que introduce una posible clasificación errónea de los participantes, datos faltantes y variabilidad en la definición de cohortes de casos y controles 8 . Identificar individuos que, de hecho, pueden ser o convertirse en casos pero que no están codificados correctamente o aún no han sido diagnosticados clínicamente (es decir, «casos crípticos») sigue siendo un desafío y una oportunidad abiertos.
En este trabajo, presentamos un enfoque sistemático (MILTON) que tiene como objetivo predecir enfermedades utilizando biomarcadores clínicos de medición común, niveles de proteínas plasmáticas y otros rasgos cuantitativos. Descubrimos que estos modelos predijeron una variedad de enfermedades diferentes con gran precisión. Estos resultados no solo brindan biomarcadores candidatos y combinaciones de biomarcadores, sino que también ayudan claramente a aumentar y, por lo tanto, a potenciar los análisis de casos y controles en algunos entornos de enfermedades. Utilizando estas predicciones validadas, realizamos estudios de asociación de fenomas (PheWAS) a nivel de genes y variantes aumentados 8 en 3213 fenotipos y 484 230 genomas UKB 9 . Esto identificó varias asociaciones gen-fenotipo supuestamente novedosas que no alcanzaron significancia en el PheWAS inicial de la misma cohorte de prueba.
Resultados
Descripción general
Los biomarcadores clínicos tienen un papel clave en el diagnóstico y la evaluación de muchas enfermedades, ya que proporcionan indicaciones mensurables de la presencia y/o gravedad de una afección. En el contexto de los PheWAS, también brindan la oportunidad de identificar casos crípticos o mal clasificados. Aquí, presentamos un método de aprendizaje automático, MILTON, que utiliza biomarcadores cuantitativos para predecir el estado de la enfermedad para 3213 fenotipos de enfermedades (Fig. 1 y Métodos ). MILTON primero aprende una firma específica de la enfermedad dado un conjunto de pacientes ya diagnosticados y luego predice nuevos casos putativos entre los controles originales (Fig. 1 y Métodos ). Las cohortes aumentadas se utilizan para repetir el análisis de colapso de variantes raras 4 y comparar los resultados con las cohortes de referencia utilizadas para entrenar los modelos (Fig. 1 ).

Definición de modelos basados en fechas de toma de muestras y diagnóstico
En el UKB, las muestras para la medición de biomarcadores pueden haber sido recolectadas hasta ~16,5 años antes o 50 años después de que el individuo correspondiente fuera diagnosticado con una enfermedad (Fig. 2a ). Para determinar el efecto de este desfase temporal en el rendimiento predictivo, entrenamos modelos MILTON en casos seleccionados de acuerdo con tres modelos temporales diferentes: pronóstico, diagnóstico y agnóstico en el tiempo (Fig. 2a ), y cinco desfases temporales diferentes ( Métodos ). El modelo pronóstico utiliza a todos los individuos que recibieron un diagnóstico hasta 10 años después de la recolección de la muestra del biomarcador; el modelo diagnóstico utiliza a todos los individuos que recibieron un diagnóstico hasta 10 años antes de la recolección de la muestra del biomarcador; y el modelo agnóstico en el tiempo utiliza a todos los individuos diagnosticados para el desarrollo del modelo ( Métodos ). Observamos que para el modelo pronóstico, los diagnósticos faltantes pueden atribuirse a que la enfermedad aún no está presente/diagnosticada o a que faltan registros en el biobanco. El punto de corte de 10 años se seleccionó como óptimo después de un análisis de sensibilidad de biomarcadores sobre el efecto de la recolección de muestras y el retraso en el diagnóstico en 400 códigos CIE10 (Clasificación Internacional de Enfermedades, 10.ª revisión 10 ) seleccionados al azar ( Métodos y Figura complementaria 1 ).
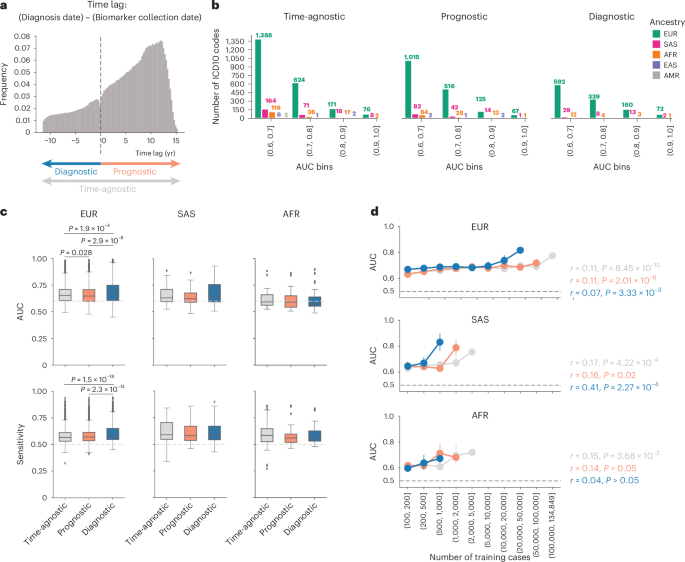
Rendimiento de predicción de la enfermedad de MILTON
En primera instancia, se entrenó a MILTON utilizando 67 características, incluidas 30 medidas de bioquímica sanguínea, 20 medidas de hemograma, cuatro medidas de análisis de orina, tres medidas de espirometría, cuatro medidas de tamaño corporal, tres medidas de presión arterial, sexo, edad y tiempo de ayuno. Después de ejecutar MILTON en el fenoma y en múltiples ancestros por separado (Tabla 1 ), hubo 3200, 2423 y 1549 códigos ICD10 basados en modelos independientes del tiempo, pronóstico y diagnóstico, respectivamente, que cumplieron con nuestro conjunto mínimo de criterios de robustez (Tabla complementaria 1b ). Utilizando el área bajo la curva (AUC) para evaluar el rendimiento del modelo, MILTON logró un AUC ≥ 0,7 para 1091 códigos ICD10, un AUC ≥ 0,8 para 384 códigos ICD10 y un AUC ≥ 0,9 para 121 códigos ICD10 en todos los modelos temporales y ancestros (Fig. 2b , Tabla complementaria 2a–e y Notas complementarias ). MILTON logró un AUC > 0,6 para más de la mitad de los códigos ICD10 estudiados por capítulo ICD10 en 13 de los 18 capítulos y los tres modelos temporales para individuos de ascendencia europea (EUR) (Fig. complementaria 2a ).
Descubrimos que los modelos de diagnóstico generalmente tuvieron un mayor rendimiento en 1466 códigos ICD10, con resultados disponibles para los tres modelos de tiempo en participantes con ascendencia EUR (AUC diagnóstica mediana versus AUC pronóstica : 0,668 versus 0,647, prueba U de Mann-Whitney (MWU) bilateral P = 2,86 × 10 −8 ; sensibilidad mediana diagnóstica versus sensibilidad pronóstica : 0,586 versus 0,570, MWU bilateral P = 2,31 × 10 −14 ; Fig. 2c y Tabla complementaria 2a–e ). En general, a medida que aumentó el número de casos disponibles para entrenamiento según CIE10, el AUC, la sensibilidad y la especificidad se mantuvieron estables para las ascendencias EUR y africanas (AFR), mientras que aumentaron para los modelos de diagnóstico del sur de Asia (SAS) (AUC: r de Pearson = 0,41, P = 2,27 × 10 −4 ; sensibilidad: r de Pearson = 0,48, P = 1,29 × 10 −5 ; especificidad: r de Pearson = 0,31, P = 0,0065; Fig. 2d y Fig. suplementaria 2b ).
MILTON predice con éxito la enfermedad antes de su aparición
Para evaluar la eficacia de MILTON en la predicción de casos genuinos, buscamos determinar si los individuos a los que se les asignó una alta probabilidad de caso (0,7 ≤ P caso ≤ 1) según MILTON en el modelo de pronóstico fueron finalmente diagnosticados con esos códigos ICD10 en actualizaciones posteriores del fenotipo UKB. Para investigar esto, entrenamos los modelos MILTON únicamente en casos diagnosticados antes del 1 de enero de 2018 y analizamos los puntajes de probabilidad predichos para los casos diagnosticados después de esta fecha (análisis limitado, Fig. 3a ).
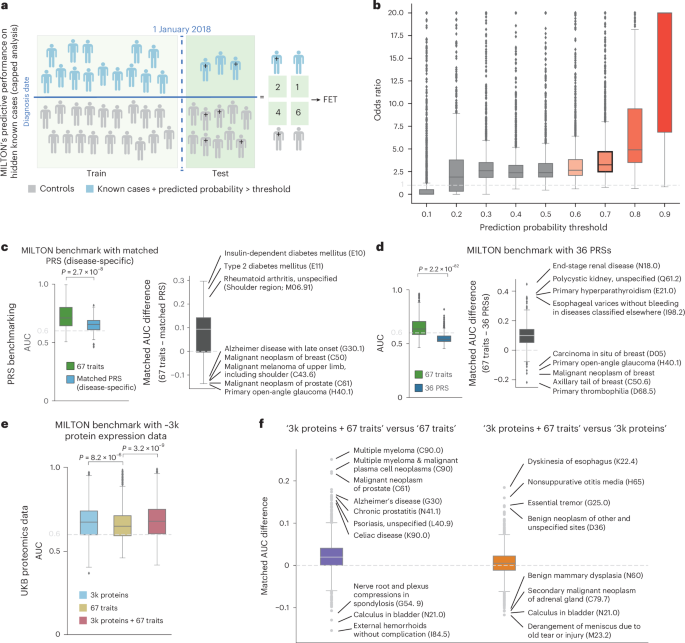
Entre 1.740 códigos ICD10 con un mínimo de 30 individuos diagnosticados después del 1 de enero de 2018, y AUC ≥ 0,6 (Tablas suplementarias 1b y 2f ), 1.695 códigos (97,41%) se enriquecieron significativamente en participantes que tenían un caso P ≥ 0,7. Esta observación fue apoyada por una razón de probabilidades mayor que 1 (prueba exacta de Fisher unilateral P < 0,05) que persistió a través de los umbrales de probabilidad de predicción ≥ 0,3 (Fig. 3b y Tabla suplementaria 3 ). Estos resultados validan la capacidad de MILTON para predecir casos emergentes a partir de un grupo de participantes no diagnosticados en ese momento, lo que enfatiza su valor para la predicción del riesgo de enfermedad y su potencial para aumentar las etiquetas de casos positivos existentes para los análisis de asociación genética.
MILTON supera las puntuaciones de riesgo poligénico para la predicción de enfermedades
Las puntuaciones de riesgo poligénico (PRS) se investigan ampliamente para ayudar potencialmente al diagnóstico de enfermedades en las clínicas 11 , 12 . Comparamos el rendimiento de los modelos MILTON entrenados en 67 rasgos cuantitativos ( Métodos ) con aquellos entrenados en la PRS específica de la enfermedad respectiva 13 de un fenotipo dado, o las 36 PRS estándar disponibles en el UKB. Usamos las 36 PRS aquí para considerar también los efectos de las PRS no específicas de la enfermedad, como las PRS para el colesterol total, las PRS para la altura, etc., en la predicción de varias enfermedades.
Observamos que los modelos agnósticos en el tiempo de MILTON entrenados en 67 rasgos cuantitativos superaron significativamente a los entrenados en un solo PRS específico de la enfermedad (incluyendo sexo y edad como covariables) para 111 de 151 códigos ICD10 (AUC media de 67 rasgos frente a AUC PRS específico de la enfermedad : 0,71 frente a 0,66, MWU bilateral P = 2,71 × 10 −8 ; Tabla 1 , Fig. 3c , Fig. 3a suplementaria y Tabla 4a suplementaria ). Observamos la misma tendencia para los modelos de pronóstico y diagnóstico ( Notas suplementarias ). Cabe destacar que las PRS para cáncer de mama (C50), melanoma (C43, D03) y cáncer de próstata (C61) tuvieron un mejor desempeño que los 67 rasgos cuantitativos en los tres modelos temporales (Fig. 3a suplementaria ), probablemente debido a que los biomarcadores basados en sangre y orina utilizados por MILTON tienen valores predictivos menores para estos cánceres sólidos. En un análisis adicional, entrenamos modelos MILTON que incluían las 36 PRS estándar proporcionadas en el UKB y nuevamente observamos que los modelos entrenados en los 67 rasgos superaron significativamente a los entrenados en PRS para 499 códigos ICD10 seleccionados al azar (AUC media de 67 rasgos frente a AUC de todas las PRS : 0,64 frente a 0,54, MWU bilateral P = 2,17 × 10 −82 ; Tabla 1 , Fig. 3d , Fig. 3c suplementaria y Tabla 4b suplementaria ).
Los datos de proteómica plasmática mejoran el rendimiento en varias enfermedades
Además de los biomarcadores clínicos estándar, la disponibilidad de otras modalidades ómicas proporciona características adicionales para predecir casos. Recientemente, el consorcio UKB Pharma Proteomics Project perfiló 2.923 proteínas plasmáticas en un subconjunto de 49.736 participantes de UKB 14 , 15 . Utilizando el subconjunto de ascendencia EUR ( n = 46.327) de la cohorte UKB, reentrenamos MILTON incorporando los datos proteómicos, tanto de forma aislada como en combinación con los otros 67 biomarcadores ya analizados ( Notas complementarias ). Esto condujo a un rendimiento general ligeramente mejorado (AUC media de proteínas 3k + 67 rasgos frente a AUC de 67 rasgos : 0,68 frente a 0,65, MWU bilateral P = 3,24 × 10 −9 ; Tabla 1 , Fig. 3e,f y Tabla complementaria 4c–e ), con 52 fenotipos que tuvieron una mejora del AUC de ≥ 0,1 (Fig. 3f ). Varios fenotipos se beneficiaron considerablemente de la inclusión de datos de proteómica plasmática (Fig. complementaria 3d y Notas complementarias ), incluido C90 (mieloma múltiple y neoplasias malignas de células plasmáticas), con una mejora del AUC de 0,63 a 0,85. Esto fue impulsado en gran medida por la adición de las mediciones de las proteínas del miembro 13B de la superfamilia del receptor TNF (TNFRSF13B o TACI) y del miembro 17 (TNFRSF17 o BCMA) 16 , 17 , 18 , 19 (Fig. 3f , Fig. suplementaria 4b y Tabla suplementaria 5f–h ). Del mismo modo, para C61 (cáncer de próstata), el AUC mejoró de 0,54 a 0,76 debido a la adición del antígeno conocido del cáncer de próstata (KLK3; Figs. 3f y 4c ). Otros ejemplos incluyeron G12 (atrofia muscular espinal y síndromes relacionados), con un AUC que mejoró de 0,57 a 0,70, probablemente debido a la adición de la proteína de la cadena ligera del neurofilamento (NEFL) 20 , 21 (Fig. 4c ). Estos resultados resaltan el valor de agregar características adicionales para ciertas enfermedades y, en particular, el poder de los datos proteómicos para impulsar las predicciones.
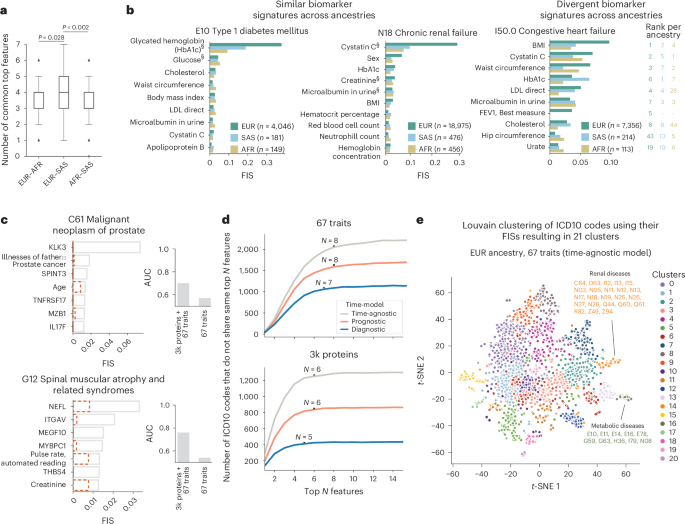
MILTON identifica características predictivas y grupos de enfermedades
MILTON nos permite inferir la importancia de cada característica en la definición de fenotipos de la enfermedad (Figs. Suplementarias 4 , 5 y 6 ), así como su concordancia entre ascendencias (Fig. 4a y Notas Suplementarias ). Observamos que MILTON asignó altos puntajes de importancia de característica (FIS) a al menos uno de los biomarcadores enumerados para el capítulo 22 de la enfermedad correspondiente (Fig. Suplementaria 6 y Tabla Suplementaria 5a–e ). Por ejemplo, la hemoglobina glucosilada (HbA1c) y la glucosa se clasificaron como las dos características principales para la diabetes mellitus tipo 1 (E10: AUC de las tres ascendencias, modelo agnóstico del tiempo = 0,93 ± 0,04; Fig. 4b ), lo que se espera ya que se utilizan para el diagnóstico clínico de la diabetes. La cistatina C, la microalbúmina en orina y la creatinina se clasificaron entre las cinco primeras en la insuficiencia renal crónica, y el sexo fue una de las principales características para predecir la insuficiencia renal crónica en todas las ascendencias 23 , 24 , 25 , 26 (ICD10: N18; Fig. 4b ). Esto indica que MILTON puede distinguir entre puntos de corte específicos para hombres y mujeres en los diferentes biomarcadores, ya que en ciertas enfermedades los rangos de referencia para algunos biomarcadores pueden ser específicos del sexo.
Observamos las características principales de aquellos códigos ICD10 en los que MILTON mostró un mejor rendimiento tras la adición de datos proteómicos (Fig. 4c y Fig. 4a,b suplementarias ) y, como validación positiva, confirmamos que MILTON clasifica a los biomarcadores que se sabe que están asociados con ciertas enfermedades como las principales características predictivas 27 , 28 , 29 , 30 (Fig. 4b,c y Fig. 4b suplementarias ). También exploramos la cantidad de características necesarias para caracterizar de forma única un fenotipo, identificando las siete u ocho características más importantes por enfermedad como suficientes para proporcionar una firma casi única para cada enfermedad (según un cambio de <5 % en la cantidad de códigos ICD10 que comparten las características N principales y N − 1 principales; Fig. 4d ). De manera similar, 5 a 6 características principales de ~3000 proteínas + covariables clínicas pueden ser suficientes para distinguir un ICD10 de otro (Fig. 4d ). Finalmente, generamos grupos de fenotipos enriquecidos con perfiles de biomarcadores similares (Fig. 4e y Tabla complementaria 6a ) que nos permiten explorar los perfiles de comorbilidad en las cohortes de pacientes (Tabla complementaria 6b y Notas complementarias ). Hemos puesto a disposición todas las características predictivas principales por enfermedad y los grupos de enfermedades en nuestro portal público ( http://milton.public.cgr.astrazeneca.com ).
El estudio PheWAS sobre cohortes aumentadas con MILTON revela supuestas señales novedosas
El poder de MILTON en la predicción del riesgo de enfermedad abre un potencial adicional: aumentar las etiquetas de casos positivos existentes para los análisis de asociación genética. Extrajimos cohortes aumentadas con MILTON (‘L0’–’L3’; Fig. Suplementaria 7a , Métodos y Notas Suplementarias ) para 2371 códigos ICD10 con AUC > 0,6 para ascendencia EUR, 271 para ascendencia SAS, 179 para ascendencia AFR, nueve para ascendencia del este asiático (EAS) y dos para ascendencia estadounidense (AMR). Aquí, las cohortes aumentadas con MILTON contienen todos los casos conocidos junto con un número creciente de casos predichos por MILTON a medida que pasamos de predicciones L0 (conservadoras) a L3 (más inclusivas) ( Métodos ). El análisis de colapso de variantes raras en genomas completos derivados de estas cohortes aumentadas de ascendencia EUR dio como resultado 2905 asociaciones significativas genes-ICD10 ( P MILTON < 1 × 10 −8 ) entre 1207 códigos ICD10 y 165 genes, el 99,93 % de los cuales tienen los valores P más bajos en modelos de variantes calificativas (QV) 4 no sinónimas (Tabla complementaria 7a ).
Para comparar los resultados de MILTON con un conjunto de datos de referencia, realizamos un análisis binario de PheWAS en las cohortes iniciales para cada código ICD10 y recuperamos 236 de 270 asociaciones gen-enfermedad de los análisis iniciales en las cohortes aumentadas con la misma dirección del efecto ( P MILTON , P basal < 1 × 10 −8 ), etiquetándolas como ‘binarias conocidas’ (Tabla 2 , Fig. suplementaria 7b y Notas suplementarias ). Para varias señales conocidas, evaluadas como controles positivos 31 , 32 , 33 , 34 , 35 , 36 , 37 , MILTON logró resultados mejorados de PheWAS (Fig. 5a y Notas suplementarias ). Al caracterizar señales nuevas putativas, buscamos detectar señales que puedan reflejar una correlación con un biomarcador en lugar de una asociación independiente con la enfermedad. Para ello, consideramos todos los genes que alcanzan significancia a nivel de todo el genoma con un biomarcador en una PheWAS cuantitativa de referencia y evaluamos si se encuentran entre los principales predictores de un fenotipo determinado en las cohortes MILTON ( Métodos y notas complementarias ). Estos representaron 1047 asociaciones significativas entre genes y enfermedades que no consideramos como supuestamente novedosas ( Notas complementarias ).
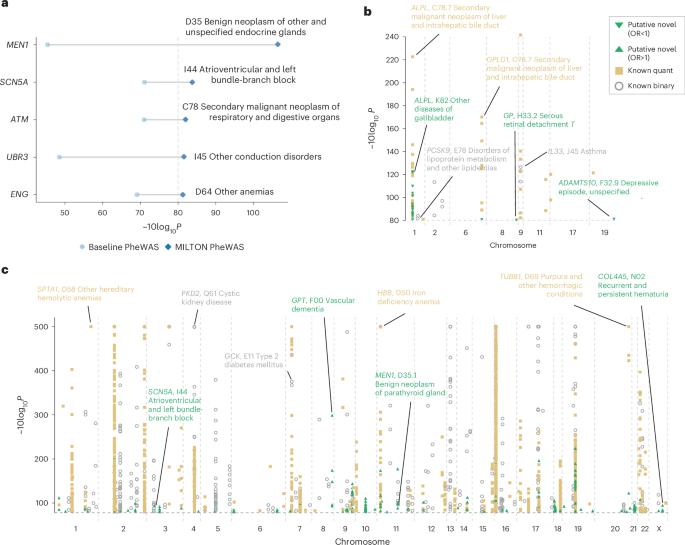
Finalmente, etiquetamos las 182 asociaciones gen-enfermedad restantes ( P MILTON < 1 × 10 −8 , P basal > 1 × 10 −8 y P PheWAS cuantitativo para FIZ > 1,2 > 1 × 10 −8 ; Tabla 2 ) como ‘novedosas putativas’ (Fig. 5b,c y Notas suplementarias ). FIZ se refiere a la puntuación Z de importancia de la característica de biomarcadores altamente predictivos por código ICD10. Para caracterizar una asociación como ‘novedosa putativa’, requerimos que para cualquier biomarcador que alcance FIZ > 1,2 durante el entrenamiento MILTON (para un código ICD10 particular), las respectivas asociaciones PheWAS cuantitativas no sean significativas ( P PheWAS cuantitativo > 1 × 10 − 8 ). Este filtrado se aplica para garantizar que cualquier señal nueva putativa no refleje correlaciones con biomarcadores sino que represente asociaciones de enfermedades independientes ( Notas suplementarias ). En general, MILTON informó 231 nuevos supuestos resultados en todos los códigos ICD10 ( Tabla 2 ), el 76,37% de los cuales también alcanzaron significancia nominal ( P < 0,05) en los PheWAS iniciales. Estos nuevos supuestos resultados alcanzaron valores P significativamente más bajos en las cohortes iniciales en todos los fenotipos en comparación con los genes seleccionados aleatoriamente del mismo tamaño ( Fig. suplementaria 8 ). Como observación de control positivo, los capítulos XXI (servicios de salud) y XVIII (hallazgos de laboratorio) se clasificaron como los principales capítulos con nuevas asociaciones putativas, ya que capturan inherentemente a individuos con niveles de biomarcadores atípicos, que es el enfoque principal de MILTON ( Fig. suplementaria 7g ).
Luego aplicamos PheWAS a ancestros no EUR, que representan una proporción menor de UKB, y observamos asociaciones significativas a nivel de genoma solo en ancestros SAS (ocho asociaciones entre tres genes y ocho códigos ICD10; Tabla complementaria 7b ). Una asociación gen-fenotipo fue compartida entre EUR y SAS, que fue la asociación basal de la subunidad beta de hemoglobina ( HBB )–talasemia D56. Después de combinar los resultados binarios de PheWAS correspondientes a variantes de truncamiento de proteínas de todos los ancestros usando la prueba de Cochran–Mantel–Haenszel, identificamos nueve asociaciones en cohortes aumentadas con MILTON que alcanzaron significancia a nivel de genoma ( P < 1 × 10 −8 ) en el análisis pan-ancestro pero tuvieron P > 1 × 10 −8 en el análisis específico de ancestros EUR (Fig. complementaria 9 y Tabla complementaria 7c ). Un ejemplo es la asociación de la apolipoproteína B ( APOB ) – E05 con tirotoxicosis (hipertiroidismo) que alcanzó P = 9,02 × 10 −9 en el análisis pan-ancestro pero tuvo una significación limítrofe ( P = 1,77 × 10 −8 ) en el análisis específico de EUR. Una variante intrónica, rs12720793 , en el gen APOB también está asociada con tirotoxicosis en el FinnGen Biobank ( P < 0,008). Por el contrario, identificamos 33 asociaciones en cohortes basales y 274 asociaciones en cohortes aumentadas que fueron significativas a nivel de todo el genoma en el análisis específico de EUR pero no en el análisis pan-ancestro (Fig. Suplementaria 9 y Tabla Suplementaria 7c ).
Estudios de asociación de todo el exoma en cohortes aumentadas con MILTON
Para evaluar si existe un beneficio adicional al usar cohortes extendidas de MILTON para estudios de asociación de variantes comunes, aplicamos MILTON al análisis de enriquecimiento a nivel de variante (estudio de asociación de todo el exoma (ExWAS)) en todos los fenotipos ( n = 2259 con AUC > 0,6), empleando nuestra línea de trabajo ExWAS de trabajo previo 4 . Con base en el modelo alélico ExWAS, observamos que la razón de probabilidades para >99% de asociaciones de variante-ICD10 ( P < 0,05) permaneció consistente en la dirección del efecto entre las cohortes MILTON y las cohortes basales (Fig. Suplementaria 12a ). El porcentaje de asociaciones con valores P potenciados en comparación con el valor basal aumentó de las cohortes L0 a L3 (53,94% a 65,25%; Fig. Suplementaria 12b ), al igual que el número de asociaciones significativas a nivel de genoma (Fig. Suplementaria 12c ). En general, recuperamos con MILTON 6.321 de 8.013 asociaciones basales a nivel de variante (78,9 %; P < 1 × 10 −8 ) y observamos 15.490 «cuantitativos conocidos» junto con 9.882 asociaciones nuevas putativas (Fig. 6c , Fig. 13 suplementaria y Tabla suplementaria 7d ). Entre las 9.882 asociaciones nuevas putativas de ExWAS, el 61,94 % ( P = 6.121) alcanzó P < 0,05 en las cohortes basales (Fig. 6c ). Para lograr una mayor granularidad, también realizamos estudios de asociación de variantes comunes del genoma completo (GWAS) en 20 códigos ICD10 y para cada una de las cohortes aumentadas con MILTON, así como las de referencia, lo que confirma nuevamente que logramos un buen enriquecimiento de casos reales en las cohortes MILTON (Tabla complementaria 9b , Métodos y Notas complementarias ).
Validación con FinnGen Biobank
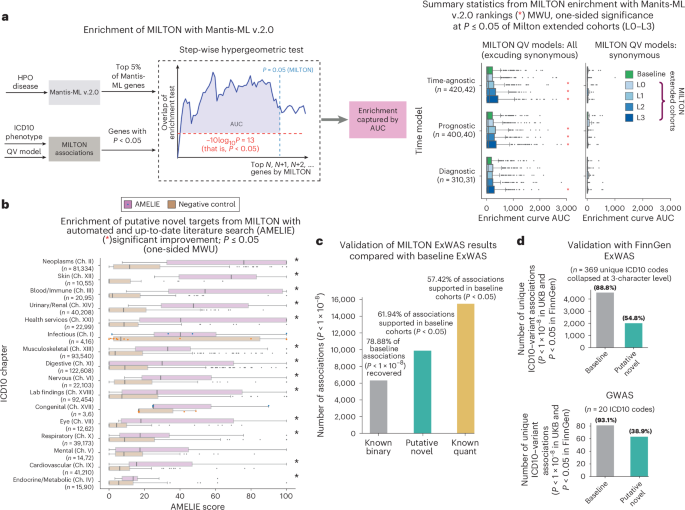
Nuestro objetivo fue identificar el respaldo a los supuestos nuevos hits basados en MILTON ExWAS en conjuntos de datos externos, y específicamente en el FinnGen Biobank 38 , que tiene resultados de enriquecimiento a nivel de variante disponibles ( Notas complementarias ). Entre todas las supuestas asociaciones de MILTON con nuevos ExWAS que se pueden probar en FinnGen, el 54,76 % ( n = 2002) alcanzó P < 0,05 en FinnGen 38 versión 10 (Fig. 6d y Tabla complementaria 9a ). Como referencia, entre los hits significativos de todo el genoma inferidos en los PheWAS basales, el 88,76 % ( n = 4525) tuvo evidencia de respaldo en FinnGen ( P < 0,05; Fig. 6d ).
Para la validación de las asociaciones de variantes comunes con la CIE10 obtenidas de los GWAS, investigamos 14 códigos de la CIE10 que se pueden asignar a los resultados de las estadísticas resumidas en FinnGen Biobank 38 versión 11 (Tabla complementaria 8 ). Entre las asociaciones de referencia únicas ( P < 1 × 10 −8 ) que se pudieron probar en FinnGen, el 93,10 % ( n = 81) tuvo P < 0,05 en FinnGen con la misma dirección del efecto (Fig. 6d y Tabla complementaria 9b ). Además, el 94,81 % ( n = 73) de las asociaciones «binarias conocidas» de MILTON tuvieron evidencia de apoyo ( P < 0,05 con la misma dirección del efecto). Entre las 167 asociaciones novedosas putativas de MILTON que se pudieron asignar a los resultados de FinnGen, el 38,92 % ( n = 65) logró P < 0,05 en FinnGen con la misma dirección del efecto (Fig. 6d y Tabla complementaria 9b ).
Los posibles nuevos éxitos ocupan un lugar destacado en dos herramientas ortogonales basadas en inteligencia artificial
Luego buscamos validar los nuevos supuestos éxitos inferidos por PheWAS en cohortes aumentadas con MILTON, utilizando dos herramientas de aprendizaje automático independientes, Mantis-ML (v.2.0) 39 , 40 y AMELIE (Evaluación automática de literatura mendeliana) 41 , 42 . Mantis-ML (v.2.0) está entrenado en todo el fenoma, aprovechando recursos centrados en genes disponibles públicamente, como Human Phenotype Ontology (HPO), Open Targets y Genomics England, así como gráficos de conocimiento para asignar rangos para cada gen en el exoma humano en miles de fenotipos. Se realizaron pruebas hipergeométricas escalonadas para comparar las predicciones de Mantis-ML (v.2.0) 39 , 40 y las asociaciones identificadas por MILTON (Fig. 6a , Métodos y Notas suplementarias ). En particular, los genes identificados en las cohortes L3 para los modelos QV no sinónimos ( P < 0,05) se enriquecieron significativamente en genes de alto rango de Mantis-ML (v.2.0) en comparación con la cohorte de referencia en los tres modelos temporales (Fig. 6a ; comparación por fenotipo mostrada en la Fig. 20a suplementaria ). Además, todos los fenotipos se enriquecieron en genes de alto rango bajo el modelo QV dañino ultra raro (frecuencia de alelo menor UKB ≤ 0,005%; ref. 4 en las cohortes aumentadas con MILTON en comparación con el modelo QV sinónimo, seguido de las variantes ultra raras según el modelo de razón de tolerancia a errores (URmtr 4 ) y el modelo de variante de truncamiento de proteínas (ptv 4 ; Fig. 20b,c suplementarias ).
AMELIE (v.3.1.0) 41 , 42 realiza una búsqueda bibliográfica automatizada, integrando actualizaciones nocturnas de todo el corpus de PubMed, para estimar las clasificaciones de genes para la causalidad de la enfermedad entre un grupo de genes candidatos. AMELIE requiere que las enfermedades se organicen dentro del HPO. Por lo tanto, primero identificamos las cinco enfermedades HPO más similares semánticamente de un grupo de 17.451 fenotipos para cada código ICD10 y consultamos a AMELIE para obtener asociaciones enfermedad-gen para esas enfermedades ( Notas complementarias ). Observamos que para 13 de los 16 capítulos ICD10, los objetivos nuevos putativos ( Tabla complementaria 7a ) tuvieron una puntuación AMELIE significativamente más alta ( P < 0,05) que los controles negativos generados a través de diez muestreos de conjuntos de genes aleatorios de la misma longitud ( Fig. 6b y Fig. complementaria 21 ).
Estas validaciones resaltan el poder de MILTON para resaltar posibles señales novedosas, también respaldadas por métodos integrales de priorización de genes basados en inteligencia artificial, que aprovechan grandes cantidades de evidencia biológica de la literatura y cientos de recursos de datos públicos.
Discusión
Presentamos aquí MILTON, un marco de aprendizaje automático para la predicción de enfermedades basada en biomarcadores y multiómica, y para potenciar los estudios de casos y controles en cinco ascendencias de UKB. A pesar de que el conjunto de características empleado en este estudio es bastante amplio y diverso y no específico de la enfermedad, MILTON logró un poder predictivo relativamente alto a alto en una cantidad considerable de fenotipos (AUC > 0,7 para 1091 códigos ICD10, AUC > 0,8 para 384 códigos ICD10 y AUC > 0,9 para 121 códigos ICD10). La falta de poder predictivo en el resto de los fenotipos indica que no todos los fenotipos pueden tener una huella de biomarcador distintiva, al menos no en el conjunto de biomarcadores utilizado actualmente. Hay ciertos capítulos de enfermedades donde los modelos podrían mejorarse mediante la inclusión de características más informativas en todas las ascendencias y modelos temporales, como el capítulo II (neoplasias), el capítulo VIII (oído) y el capítulo XII (piel) (Fig. Suplementaria 2a y Tabla Suplementaria 2a–e ). Por ejemplo, la adición de datos de expresión macromolecular de marcadores ya conocidos de cáncer de mama (C50) 43 puede ayudar a mejorar el rendimiento del modelo. Es importante señalar que, si bien MILTON superó a las PRS para la mayoría de las enfermedades, para ciertas enfermedades, como el melanoma, el cáncer de mama, el cáncer de próstata y el glaucoma primario de ángulo abierto, las PRS tuvieron un mayor poder predictivo (mejora del AUC ≥ 0,1) que los 67 rasgos cuantitativos empleados por MILTON. Actualmente, utilizamos PRS estándar generados por Thompson et al. 13 en lugar de PRS mejorados 13 porque las últimas puntuaciones estaban disponibles solo para el 20,75 % ( n = 104 231) de los individuos. Tal vez los modelos entrenados con PRS mejorados o PRS derivados de otras formulaciones puedan mejorar el rendimiento, especialmente en el caso de enfermedades en las que no se dispone de biomarcadores de diagnóstico o pronóstico claros. La inclusión de datos proteómicos también ayudó a lograr un mejor rendimiento (ΔAUC ≥ 0,1) para 52 fenotipos. Por lo tanto, alentamos la exploración de otros espacios de características que podrían mejorar aún más el rendimiento de nuestros modelos en fenotipos adicionales.
En algunas ocasiones, la aplicación de MILTON redujo el poder de descubrimiento de señales al diluir las señales de genes y enfermedades del análisis de referencia, que era ingenuo para MILTON. Sin embargo, en general, el 87,41 % de las señales conocidas del análisis de referencia de PheWAS tuvieron una señal mejorada o mantuvieron su importancia a nivel de todo el genoma con la misma dirección del efecto. Por lo tanto, MILTON conduce con más frecuencia a una mejor detección de señales y rendimiento de descubrimiento que a señales disminuidas, lo que brinda una gran confianza en que este enfoque realmente está aumentando las pruebas de asociación tradicionales (de referencia) que se basan únicamente en definiciones de casos etiquetados positivamente en grandes biobancos.
Otras 182 señales eran señales de genes y enfermedades supuestamente nuevas que no fueron descubiertas ni por el análisis de casos y controles de referencia ni por el análisis cuantitativo de rasgos de PheWAS. Estas supuestas asociaciones novedosas deben validarse experimentalmente más.
Recientemente, han surgido métodos avanzados basados en aprendizaje profundo para la imputación de características 44 , que pueden ayudar a superar algunos de los desafíos recurrentes que enfrentan los conjuntos de datos de biobancos en torno a la falta de datos y la imputación. MILTON se distingue de los métodos que se centran en la imputación de características 44 de rasgos binarios o cuantitativos faltantes (es decir, de las variables independientes), y en cambio se centra en aumentar los fenotipos de enfermedad predichos (es decir, la variable dependiente) aprovechando una gran cantidad de características de entrada. Específicamente, MILTON primero aprende una firma de enfermedad, dado un conjunto de rasgos binarios y cuantitativos, y luego predice individuos no diagnosticados que comparten la firma aprendida por enfermedad, refinando efectivamente las definiciones originales de cohorte de casos y controles. Las mejoras futuras de MILTON podrían incluir una selección flexible de modelos de tiempo y desfase temporal para cada código ICD10 para reflejar la dinámica variable del desarrollo de la enfermedad y los perfiles de biomarcadores que caracterizan estos pródromos ( Notas complementarias ).
Además, MILTON ofrece una forma de mejorar el análisis de colapso de variantes raras, al tiempo que cierra la brecha entre los biomarcadores clínicos y las enfermedades. En los casos en los que los principales biomarcadores predictivos carecen actualmente de una conexión biológica conocida con una enfermedad, mientras que el modelo predictivo logra un buen rendimiento (AUC > 0,8), sugerimos que se exploren más estos vínculos a medida que nuestro conocimiento de los biomarcadores específicos de la enfermedad continúa creciendo. Se recomienda a los expertos que tengan cuidado al analizar los estudios de asociación posteriores en tales casos. Los conjuntos de biomarcadores inferidos por MILTON pueden proporcionar información para definir conjuntos mínimos de biomarcadores, independientemente del diagnóstico, que se recopilarán como parte de futuros biobancos y se pondrán a disposición de los investigadores registrados. Finalmente, MILTON proporciona el poder de caracterizar conjuntos de características basadas en la sangre específicas de la enfermedad que pueden definir y predecir una amplia gama de patologías humanas y proporcionar posibles conocimientos relacionados. Esto tiene implicaciones para futuras estrategias de prevención y detección temprana de enfermedades.
