Dr. Carlos Alberto Díaz.
Comentarios vinculados a una búsqueda bibliográfica.
Importancia de la intervención conceptual:
A medida que surgen nuevos datos y evidencia clínica sobre esta enfermedad, la naturaleza vascular de COVID-19 se ha centrado. Comprender la cascada desregulada clave desencadenada por COVID-19 en el sistema vascular nos proporciona un marco para informar las intervenciones sobre los mediadores clave del proceso patólogo, y de esta manera reducir la morbilidad y mortalidad a corto y largo plazo de esta enfermedad pandémica.
En salud, el endotelio vascular mantiene la homeostasis a través de la regulación de la competencia inmune, equilibrio inflamatorio, barreras unión estrechas, estabilidad hemodinámica, así como vías trombóticas y fibrinolíticas óptimamente equilibradas.
En la nueva enfermedad del coronavirus de 2019 (COVID-19) causada por el coronavirus 2 del síndrome respiratorio agudo grave (SARS-CoV-2), la desregulación de muchas de estas vías ha surgido como mediador de la enfermedad grave.
Esto provoca una constelación de trastornos clínicos y biomarcadores observados en COVID-19 se puede clasificar en la interrupción de la competencia inmune, afectación del sistema renina-angiotensina-aldosterona (RAA), y el equilibrio trombótico, todos los cuales convergen en el endotelio vascular como una vía común.
La acumulación de evidencia en ciencia básica, imágenes y observaciones clínicas, ha aclarado el panorama de COVID-19 como una enfermedad vascular. Comprender la enfermedad en este contexto puede proporcionar nuevas vías de comprensión de COVID-19 y conducir a mejoras críticamente necesarias en las estrategias terapéuticas.
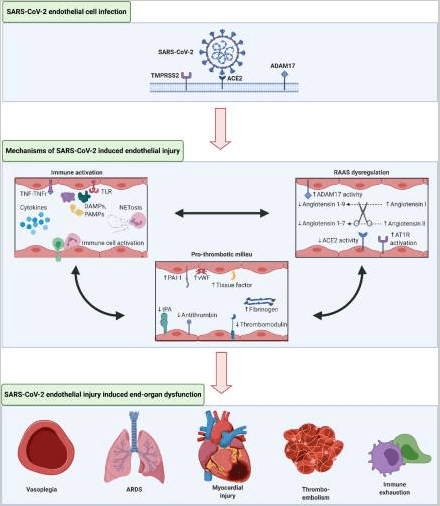
El SARS-CoV-2 utiliza la enzima convertidora de angiotensina 2 en su receptor (ACE2) para facilitar la entrada en las células diana e iniciar la infección. Esta entrada viral en la célula es mediada además por la proteasa de serina transmembrana 2 (TMPRSS2) y la catepsina L que cortan la proteína S en la partícula viral para permitir la unión con ACE2.
Las células endoteliales (CE) en general y los miocitos cardíacos en particular expresan abundantes ACE2, convirtiéndolas en un objetivo directo de la infección por SARS-CoV-2 (1 ) [2]. El examen del lecho vascular pulmonar muestra graves trastornos en COVID-19, en comparación con los pacientes con control y con gripe, particularmente con trombosis y microangiopatía generalizadas, activación endotelial y angiogénesis extensa [3]. Estos estudios y hallazgos generalizados establecen el papel de la lesión viral en el sistema vascular con la disfunción vascular resultante en pacientes con COVID-19 [4].
El endotelio vascular tiene un papel intrincado en la regulación inmune y la inflamación, es un eje que la infección SARS-CoV-2 perturba. Los informes de pacientes hospitalizados con COVID-19 revelan una activación del sistema inmunitario que muestra hallazgos consistentes con la tormenta de citoquinas, el síndrome activador de macrófagos y el posterior agotamiento inmune, el más grave en los pacientes más enfermos [5,6]. Este estado hiperinflamatorio tiene efectos nocivos en el sistema vascular con la disfunción EC resultante. En presencia de mediadores inflamatorios circulantes como la interleucina (IL)-1, IL-6, los patrones moleculares asociados al daño y los patrones moleculares asociados a patógenos, las ECs pasan por una transición a un estado activado que participa en las defensas del huésped [7]. Los IC activados promueven la inflamación localizada induciendo la expresión génica proinflamatoria, atrayendo las células inmunitarias, promoviendo el reclutamiento de células inflamatorias a tejidos lesionados o infectados, la fuga vascular aumentando la permeabilidad endotelial y alterando el potencial trombótico de la superficie local intimal. La activación de los neutrófilos conduce a la formación de trampas extracelulares de neutrófilos (NET), un proceso a veces conocido como NETosis, que puede contribuir a las respuestas a patógenos y trombosis [8]. Además, la lesión de la CE se ve agravada por la activación de receptores similares a peajes (TLR) mediante el reconocimiento de ARN viral, con el aumento de la producción de especies oxidativas reactivas (ROS) [9].
En una respuesta inmunitaria regulada y auto-limitada, estos mecanismos ayudan a sofocar el insulto local, con la curación posterior y volver a un estado de la CE en reposo. Por el contrario, los estados de una mayor respuesta inmunitaria innata y un estado protrombótico provocados por mediadores inmunes innatos como el observado en COVID-19 conducen a una cascada creciente de estas vías que promueven una profunda disfunción micro y macrovascular de la CE y daños, con el deterioro de otras funciones importantes del endotelio (1). Informes recientes de un síndrome hiperinflamatorio similar al síndrome de shock de la enfermedad de Kawasaki en niños, con biomarcadores elevados de lesión cardíaca, desarrollo de arritmia y progresión a aneurismas coronarios gigantes en algunos casos, arrojan luz sobre una manifestación de un fenómeno inmunitario centrado en macrovasculares en COVID-19, un área que justifica un estudio adicional.
Las ECs pulmonares tienen un papel importante en la vigilancia inmune, manteniendo la integridad alveolar y asegurando el intercambio adecuado de oxígeno. En COVID-19, la infección viral directa y la respuesta inflamatoria sistémica probablemente conducen a una disfunción grave de estas importantes ECs, lo que agrava el panorama resultante de la hipoxia grave y el síndrome de dificultad respiratoria aguda notificados con frecuencia en pacientes hospitalizados.
El endotelio, junto con sus funciones inmunorreguladoras clave, también desempeña un papel esencial en el mantenimiento de una interacción dinámica entre los factores pro-coagulantes y fibrinolíticos en el sistema vascular. En estado de reposo, el endotelio forma una barrera entre la capa subendotelial protrombótica y los factores pro-coagulantes en la sangre
En tiempos de estrés, los ECs activados expresan más inhibidor del activador de plasminógeno-1 (un inhibidor clave de la fibrinólisis endógena), factor tisular (un potente procoagulant) y liberan el factor von Willebrand (una proteína que forma multiméos que promueven el crecimiento del trombo). Las ECs activadas reducen la actividad de trombomombina y activador de plasminógenos tisulares, favoreciendo la acumulación de trombos [11]. Además, durante el estrés, los efectos inmunes potencian la respuesta trombótica, incluida la formación neta, y por lo tanto también pueden propagar la trombosis [8]. Estos cambios, junto con otras vías reguladoras complejas conducen a la disfunción endotelial, la activación de la cascada de coagulación y la supresión de los mecanismos fibrinolíticos, con un ambiente protrombótico resultante (1) [11]. Tales alteraciones en el equilibrio fino de la salud endotelial y los sistemas de coagulación y fibrinolítico conducen a la enfermedad trombótica microvascular macrovascular y difusa en el paciente [12].
Los estudios clínicos muestran que los pacientes con COVID-19 han aumentado el fibrinógeno, los productos de degradación de la fibrina, el factor D-dimer y von Willebrand, y estas elevaciones parecen correlacionarse con la gravedad de la enfermedad y el riesgo trombótico [13,14]. Los primeros informes mostraron una carga sustancial de lesión miocárdica en pacientes que estaban gravemente enfermos o murieron de COVID-19 [15]. Múltiples mecanismos pueden causar lesiones cardíacas en COVID-19 [16]. Si bien la infección y las tensiones hemodinámicas de la enfermedad crítica aguda pueden desencadenar la ruptura de la placa y el infarto de miocardio resultante, informes recientes indican que algunos pacientes con COVID-19 muestran hallazgos biomarcadores y electrocardiográficos de infarto de miocardio sin evidencia de ruptura aguda de la placa en la angiografía
Los datos obstétricos recientes también añaden evidencia a que SARS-CoV-2 actúa a través de mecanismos endoteliales y vasculares. En una serie de casos de 42 embarazos admitidos con infección por SARS-CoV-2, el 14% de todas las mujeres y el 75% de las mujeres con síntomas graves de COVID-19 manifestaron una constelación de síntomas similares a la preeclampsia y la hemólisis, enzimas hepáticas elevadas, síndrome de recuento bajo de plaquetas (HELLP), con resolución espontánea después de la recuperación de COVID-19 [25]. La preeclampsia tiene una disfunción vascular y endotelial considerable en su patobiología central, y el desarrollo de este trastorno a un ritmo considerable en mujeres con COVID-19 sugiere la afectación endotelial como secuela de infección viral.
El sistema de renina-angiotensina-aldosterona (RAA) que regula crucialmente la salud vascular y la función también participa en COVID-19. Como ACE2 contribuye críticamente a la biología de la infección por SARS-CoV-2, mucha atención se ha centrado en la interacción entre COVID-19 y el sistema RAA. La angiotensina II es el principal efector vascular del sistema RAA y ejerce sus efectos nocivos sobre el sistema cardiovascular a través de receptores de angiotensina-II tipo 1 (AT1) mediante la activación de vías vasoconstrictoras, inflamatorias y fibrosas. Estudios previos muestran que la angiotensina II perturba las funciones endoteliales de múltiples maneras, incluyendo el reclutamiento de monocitos, la formación de ROS, la activación de vías proinflamatorias, incluyendo a través del factor nuclear kappa-light-chain-enhancer de la regulación de las células B activadas (NF–B), así como la promoción de la producción del inhibidor activador de plasminógeno-1 (PAI-1) en las CE [26,27]. En salud, ACE2 desempeña un papel contrarregulación en el sistema RAA al cortar la angiotensina II a la angiotensina 1-7. La enzima también actúa sobre la angiotensina I para convertirla en angiotensina 1-9. Ambas conversiones enzimáticas producen péptidos que tienen potentes propiedades antiinflamatorias, antioxidantes y anti fibromáticas.
Los primeros datos indican que SARS-CoV-2 limita la expresión de ACE2 promoviendo la escisión ACE2 por la proteína especializada A desintegrina y metaloproteinasa 17 (ADAM17) y el desprendimiento de la superficie celular, lo que conduce a una reducción del papel protector de ACE2 en las CE y otros órganos [28]. Un estudio en pacientes con COVID-19 mostró un nivel notablemente elevado de angiotensina sérica II, lo que implica aún más un sistema ARA activado en las manifestaciones de la infección por SARS-CoV-2 [29]. Un mecanismo por el cual la infección por SARS-CoV-2 puede afectar la disfunción vascular es a través del estrés oxidativo. Nox2, regulado por la angiotensina-II contribuye al estrés oxidativo en el endotelio a través de la producción de especies oxidantes reactivas, y aumenta en COVID-19 grave, lo que aumenta los posibles efectos nocivos de la desregulación de la RAA en la infección por SARS-CoV-2 [30]. En general, la infección por SARS-CoV-2 da como resultado la activación del sistema RAA con su perfil resultante de las funciones endoteliales perjudiciales (1). Esta desregulación potencia la estimulación inmune innata, el estrés oxidativo y los estados protrombóticos descritos anteriormente y alimenta un círculo vicioso en la enfermedad vascular de COVID-19 [26].
El hallazgo de que D-dimer se relaciona con la gravedad de la enfermedad y la inflamación no es sorprendente dada la comprensión evolutiva de la interacción entre la inflamación y la activación de la coagulación. Algunos pacientes parecen tener una respuesta inflamatoria más pronunciada a la infección por SARS-CoV-2, con manifestaciones de un síndrome de respuesta inflamatoria sistémica (SIRS) o tormenta de citoquinas. Esta perspectiva puede explicar la variabilidad en las respuestas hipercoagulables y la variabilidad de las pruebas de coagulación, incluyendo D-dimer significativamente elevado, especialmente a medida que la enfermedad progresa [24].
Los médicos se enfrentan a un patógeno cuyo comportamiento continúa definiéndose y están buscando desesperadamente tratamientos que puedan mejorar los resultados de los pacientes [5]. COVID-19 se asocia con una lesión endotelial vascular dramática.
Es la falta de inmunidad al SARS-CoV-2 lo que sigue impulsando tanto la infección como el asombroso número de pacientes, así como la gravedad de la enfermedad, con aumentos continuos en todo el mundo. En este momento en que las vacunas se encuentran en las primeras etapas de administración con una eficacia aún indeterminada, y sin inmunidad adquirida previamente durante algún tiempo, los médicos deben seguir centrándose en la lesión endotelial vascular que se produce y evaluar posibles intervenciones terapéuticas
