Este es un trabajo colaborativo de gestión Lean Healthcare, realizado en Italia, por Desai Antonio. Goreti Giulia Giordano Mauro. Voza Antonio. para mostrar el empleo en la emergencia- pronto socorro de esta metodología, que constituye una de las categorías que he elegido para publicar sistemáticamente durante estos dos últimos años, y me pareció un abordaje y la iniciativa un proceso PDCA, disruptivo, interesante, práctico y de sentido común. Es un método para mejorar la calidad, la seguridad, la eficiencia, eliminar los desperdicios, es una filosofía comprometida con los procesos asistenciales con el aprendizaje de todos los profesionales de esas oportunidades de mejoras, conjuga un conjunto de herramientas, con el compromiso del equipo de trabajo, enfocados en la línea de atención, definir lo que no agrega valor en relación a lo que queremos, los desperdicios y las mudas, el primero de los desperdicios es sobre producir, más de lo que el proceso que sigue tolera, o es capaz de continuar, esto desequilibra el proceso que sigue. Tener exceso de inventario, más de lo que necesitamos, y luego no encontramos, ergonómicamente más difícil de manejar. Las esperas es el prínceps de las mudas en sanidad. Nuestros pacientes hacen un pase y luego espera. Juntar lotes. Movimiento inútil del personal, buscando cosas que se quedan escondidas donde no deberían estar. Transporte innecesario de los pacientes, para ello hay que diseñar el flujo correcto de las estaciones. Sobreproceso hacer más de lo que el standard dice. Estudios de laboratorio. Por último, los errores por mala calidad. Finalmente, no escuchar las iniciativas de los trabajadores. Debemos intentar eliminarnos para que los proceso sean más rápidos y con menos errores. Para ello los agentes, compradores de información y logística soliciten lo necesario, hagan bien lo que hacen con más seguridad, los proveedores internos, de los servicios deben proveer adecuadamente más rápido. Implicando al personal, desarrollo de los integrantes para el cambio, compromiso y respeto y búsqueda de la perfección. Dibujar el proceso, con la participación de los profesionales, identificando dónde pueden estar los desperdicios, si no podemos aflorar los problemas no podemos solucionar. Si no tienes ningún problema estamos en un problema. Utilizando el ciclo de Deming dé PDCA identificar bien el problema, analizar la causa raíz y desarrollamos soluciones, la implantamos, las llevamos a cabo, las midamos y estandaricemos para que se incorpore a la cultura. Los PDCA son múltiples y periódicos. Entender cómo se produjeron las mejoras, cómo se sostienen y ver la situación futura ideal de nuestros procesos, implementando los nuevos estándares, llevar a los tiempos de espera y la duración del ciclo completo. Siempre en las capacitaciones surge la pregunta y cómo empezar: se comienza con la gestión visual, medir y mejorar, y luego utilizar el sentido común, orden y limpieza, comprometer a las personas sensibilizándolas con el cambio y motivando que lo hagan por sus talentos..
El sistema de salud y la organización interna del hospital debían resolver al menos cuatro problemas, a saber, contrarrestar el contagio en la población, ofrecer el mejor tratamiento médico e intensivo a los pacientes infectados, limitar la tasa de contagio entre los trabajadores de la salud y mantener el nivel de atención adecuada de otras patologías. La búsqueda compulsiva del modelo más adecuado para cumplir con estos cuatro requisitos principales llevó a hipotetizar una ventaja teórica del método Lean.
La filosofía Lean se ha difundido ampliamente en la atención médica para reducir el desperdicio, mejorar la calidad y la eficiencia de la atención y garantizar la seguridad del paciente.Un trabajo reciente revisó sistemáticamente una aplicación de gestión ajustada en el Departamento de Emergencias, destacando los múltiples beneficios de su adopción reportados en la literatura [ 1 , 2].
En particular, lean ayuda a superar varios desafíos, como el tiempo de espera, el flujo de pacientes, la falta de disponibilidad de camas, la dificultad en la detección y la duración de la estadía en el hospital.
El estudio también reveló la importancia de la interacción entre los grupos de trabajo, que es imperativa para la implementación lean. El compromiso de los líderes con el progreso del trabajo en equipo y la cultura de mejora continua son también elementos esenciales.
Chen y sus colegas informaron una adopción lean para establecer un departamento de emergencias de fiebre de origen desconocido (FUO) que combina la operación diaria con la prevención y el control, ayudando a China y otros países a manejar el brote de enfermedades infecciosas fulminantes, a través de la implantación de tecnología de la información, la transformación local. del sitio, la asignación racional de equipos médicos y la distribución planificada de equipo de protección,3 ].
Además, otros estudios se centraron en la vía de emergencia en el período COVID 19. Patey y sus colegas informaron de una reorganización en un departamento de emergencia rural durante la enfermedad por coronavirus 2019 (COVID-19), aunque sin utilizar principios lean [ 4 , 5 ].
Los principios inspiradores del modelo operativo Lean lo convierten en un candidato ideal para el manejo de un brote de COVID-19 dentro del departamento de emergencias (SU), en comparación con los algoritmos de tratamiento estándar que no brindan rutas detalladas y específicas para los pacientes con SARS-CoV-2. .
El método Lean se realiza concretamente mediante pasos graduales y estandarizados, ahora identificables por el término “Lean-ing” [ 5 , 6 , 7 ].
La presente breve comunicación tiene como objetivo una evaluación crítica de los resultados generales de lo que comenzamos a denominar «Lean-ing» en el manejo de COVID-19 en nuestro servicio de urgencias.También se analizó la eficacia de la adopción del modelo Lean para limitar el contagio entre los trabajadores de la salud.
El algoritmo operacional Lean
La asistencia sanitaria ajustada es un «enfoque basado en herramientas». El algoritmo se basa en la implementación de técnicas y herramientas lean [ 8 ].El modelo Lean aplicado para el manejo del brote pandémico de SARS-CoV-2 abarcó la implementación de un trabajo estándar, con el objetivo de optimizar la práctica clínica y poder implementar la calidad de la atención. Todos tienen que seguir el trabajo estándar para garantizar la más alta calidad de tratamiento tanto para los pacientes como para el hospital hasta que se encuentre un enfoque más apropiado. En ese caso, se convierte en el nuevo estándar de atención.En primer lugar, se definió un protocolo interno previo al triaje (trabajo estándar). Los pacientes fueron examinados al aire libre en busca de fiebre, síntomas respiratorios y gastrointestinales para determinar la ruta más adecuada a tomar en el servicio de urgencias. Los pacientes con síntomas de SARS-CoV-2 fueron transportados directamente al hospital de las instalaciones del Hospital COVID-19, donde se sometieron a nuestro examen de primer nivel en un área de aislamiento designada. Una enfermera bien entrenada y completamente vestida con equipo de protección personal (EPP) realizó el frotis nasofaríngeo, la oximetría de pulso, tomó los signos vitales y realizó un examen físico rápido.
Los casos sospechosos se clasificaron en un área aislada específica para asignar un código de prioridad apropiado. Se prestó especial atención a la saturación de oxígeno periférico (SpO 2 %): Pacientes con una SpO 2<90% (o <86% si la enfermedad pulmonar obstructiva crónica – pacientes con EPOC) recibió inmediatamente un código 1 o 2 y se colocó en un área de tratamiento de alta intensidad; a los sujetos con una SpO 2 > 94% se les asignó un código 3 o superior y se les colocó en un área de tratamiento menos intensivo.
La zona gris estaba representada por pacientes sospechosos que mostraban un 90% <SpO 2 <94% (o 86% <SpO 2 <90% si EPOC). A estos pacientes se les administró oxígeno de bajo flujo (4 litros / min) con una cánula nasal para evaluar cualquier mejora en la SpO 2.. A los pacientes con escasa o nula mejoría en la oxigenación periférica se les asignó un código 1 o 2 y se les colocó en el área aislada mencionada anteriormente. Además, todos los pacientes recibieron una rápida evaluación médica completa, una ecografía de tórax (EE. UU.), Análisis de sangre de laboratorio y oxígeno suplementario y tratamiento médico si fuera necesario ( Figura 1 ).
En el entorno de emergencia, donde el tiempo es vital y la tasa de afluencia de pacientes es alta, el riesgo de cometer un error potencial es mayor. En la metodología Lean, el error humano puede provocar una falla en el proceso. Basado en esta suposición, lean adopta herramientas simples para disminuir la tasa de error (Poka Yoke). Dentro del área de aislamiento se colocó una pizarra para el control visual (manejo visual) de todos los pacientes con el fin de tener siempre disponible la información necesaria para el correcto manejo de los pacientes ( Figura 2 ). Además, se dibujaron símbolos en el suelo para aclarar los caminos a seguir y reducir la posibilidad de infección y errores logísticos. Además, se crearon y revisaron informes de datos y estadísticas hospitalarias a diario para mejorar los estándares de atención.
Lean tiene dos objetivos principales, a saber, la identificación y eliminación de procesos ineficientes (es decir, eliminación de desperdicios como espera, movimiento, procesamiento excesivo) y la integración de la retroalimentación de los compañeros de trabajo (multidisciplinariedad y compromiso). De gran importancia fue el sentimiento de ser parte de un grupo unido.Los trabajadores de la salud participaron en todos los niveles sin descuidar la opinión de nadie. Se realizaron reuniones multidisciplinares semanales para compartir y mejorar la gestión logística, clínica y relacional conduciendo a un aumento constante de la eficiencia y seguridad. La Figura 3 resume el enfoque Lean de COVID-19 en el servicio de urgencias.
Los datos generales de los pacientes tratados en el servicio de urgencias en el período de tiempo del 21 de febrero al 28 de mayo de 2020 se analizaron retrospectivamente.
3. Resultados operativos del sistema
Un total de 7778 pacientes ingresaron en el servicio de urgencias, de los cuales 1709 (21,9%) eran sospechosos de infección por SARS-CoV-2.El número medio de accesos diarios y casos diarios sospechosos de COVID-19 fueron 79,36 (± 23) y 17,4 (± 8), respectivamente.
Entre los casos sospechosos, 133 pacientes (7,7%) fueron asignados con un código 1, 420 (24,5%) con un código 2 y 1156 (67,6%) con un código de menor prioridad.
Después de las evaluaciones clínicas, instrumentales y de laboratorio, un total de 820 pacientes (47,9% de los casos sospechosos) cumplieron con los criterios de orientación provisionales de la OMS para el diagnóstico de SARS-CoV-2.
Murieron ciento ochenta pacientes, lo que representa una mortalidad global del 21,9%. Todos los pacientes fueron tratados con base en los mejores estándares de atención. Ciento setenta (4.8%) profesionales dieron positivo por SARS-CoV-2.
4. Discusión
En diciembre de 2019, surgió el nuevo coronavirus en Wuhan, la capital de Hubei, China. El betacoronavirus de ARN envuelto pronto se denominó SARS-CoV-2 debido a su similitud filogenética con el SARS-CoV [ 9 , 10 , 11 , 12 ]. El 20 de febrero de 2020, se detectó el primer caso del nuevo COVID-19 en Italia y, 20 días después, la OMS declaró que el brote de COVID-19 era una pandemia mundial [ 13 , 14 , 15 , 16 ]. Italia tenía una de las tasas más altas de infección por SARS-CoV-2 en todo el mundo, con 3044 casos por cada 100.000 personas, así como la segunda tasa de mortalidad más alta, 13,4% frente a un valor medio del 6,9% (al 22 de abril de 2020;https://coronavirus.jhu.edu/map.html (consultado el 13 de febrero de 2020)). La región de Lombardía, con 10,6 millones de personas que viven en zonas densamente pobladas, ha sido el epicentro de la infección, con el 37,4% del total de casos registrados en el país y más del 50% del total de defunciones ( http: // www. salute.gov.it (consultado el 13 de febrero de 2020)) [ 17 , 18 , 19 ]. La tasa de mortalidad por desarme no solo estaba relacionada con la neumonía grave, sino que también estaba muy influenciada por la afectación multiorgánica de COVID-19 [ 20]. Las comorbilidades, las complicaciones cardiovasculares, la insuficiencia renal o hepática, la respuesta hiperinflamatoria, el shock agudo y las secuelas neurológicas deben considerarse como el aspecto más insidioso e impredecible de la pandemia [ 21 , 22 , 23 , 24 , 25 , 26 , 27 , 28 , 29 , 30 , 31 , 32 , 33 , 34 , 35 , 36 , 37 , 38 ].La situación general del norte de Italia empeoró aún más por la falta de preparación del sistema de salud, como ocurre en la mayor parte del mundo. Este trágico escenario impuso la necesidad de poner en práctica un algoritmo de gestión capaz de contrarrestar la propagación de la infección, luchando al mismo tiempo contra el tiempo. A la luz de estos supuestos y al igual que para otras enfermedades en el campo de la medicina de urgencias, se necesitaba urgentemente un modelo organizativo practicado, factible, rápido y eficaz, especialmente en los Servicios de Urgencia [ 39 , 40 , 41 , 42 , 43 , 44 , 45 , 46 , 47 , 48 ].En el SU del Centro de Investigación y Clínica Humanitas, aplicamos, poco después de la declaración de la pandemia, el modelo Lean. Como lo demuestra la literatura, la implementación de Lean ofrece tremendas oportunidades en el servicio de urgencias para mejorar el hacinamiento, los costos y la pérdida de tiempo, en comparación con los modelos tradicionales [ 49 , 50 , 51 , 52 , 53 , 54 , 55 ].En 2009, Dickson et al. Evaluaron la aplicación del método Lean en el servicio de urgencias y demostraron la disminución de la duración de la estancia hospitalaria y el aumento de la mejora de las visitas de los pacientes [ 56 ]. En 2011, Holden et al. revisó la aplicación del modelo Lean en varios hospitales estadounidenses, australianos y canadienses, revelando su éxito organizacional [ 57 ]. Improta y su grupo, en noviembre de 2015, pusieron en práctica el método Lean en el servicio de urgencias del Hospital Cardarelli en Nápoles. Los datos adquiridos revelaron la eficacia de esta estrategia en la contención de costes y la seguridad del paciente [ 58 ]. Más recientemente, en 2021, Aminjarahi et al. realizó una revisión sobre la aplicación de técnicas Lean en la DE, también con el objetivo de mejorar la calidad del tratamiento [ 59].De acuerdo con los resultados de la literatura, aplicados a nuestro servicio de urgencias, el enfoque Lean parece reducir los errores y el tiempo de espera, lo que conduce a una mejora en la calidad de la atención de los pacientes. Se definió para pacientes y profesionales de la salud, priorizando el bienestar de ambos y asegurando procesos seguros, lineales y compartidos. El modelo propuesto tuvo impactos extremadamente positivos durante el brote y se destaca como uno de los pocos estudios sobre la adopción lean en los servicios de urgencias para enfrentar y reorganizar las admisiones de los pacientes durante el brote de COVID-19. Además, este trabajo contribuye a promover un algoritmo en el que se integra lean mediante un enfoque multidisciplinario [ 2 ].
Conclusiones
En nuestra experiencia, la implementación del modelo Lean ha demostrado ser eficaz para optimizar el manejo general de los casos sospechosos o determinados de pacientes con COVID-19 en un entorno de emergencia. Permitió examinar un gran volumen de pacientes y sucederlos a través de códigos de prioridad válidos. También ha sido útil para limitar la tasa de infección de los trabajadores de la salud.Si bien se admite la característica retrospectiva de nuestras observaciones, así como el número relativamente limitado de pacientes, los resultados del presente estudio presentan la experiencia de un DE en el epicentro italiano de la pandemia de COVID-19. La investigación futura se dirigirá a aplicar el modelo Lean a varias áreas de la salud para construir nuevos protocolos para la práctica clínica. La estrategia Lean puede mejorar el sistema de salud, incluso reduciendo costos.Los resultados de su aplicación práctica deben ser validados por estudios adicionales relacionados con la contención de la propagación de la infección por SARS-CoV-2 y la reducción del contagio.
D’Andreamatteo, A.; Ianni, L.; Lega, F.; Sargiacomo, M. Lean in healthcare: A comprehensive review. Health Policy2015, 119, 1197–1209. [Google Scholar] [CrossRef]
Souza, D.L.; Korzenowski, A.L.; Alvarado, M.M.; Sperafico, J.H.; Ackermann, A.E.F.; Mareth, T.; Scavarda, A.J. A systematic review on lean applications’ in emergency departments. Healthcare2021, 9, 763. [Google Scholar] [CrossRef]
Chen, T.; Ma, X.; Zhou, S.; Wang, H.; Pan, Y.; Chen, L.; Lv, H.; Lu, Y. Establishing a standardized FUO emergency department: Design and practice in dealing with COVID-19. Ann. Transl. Med.2020, 8, 749. [Google Scholar] [CrossRef]
Patey, C.; Asghari, S.; Norman, P.; Hurley, O. Redesign of a rural emergency department to prepare for the COVID-19 pandemic. CMAJ2020, 192, E518–E520. [Google Scholar] [CrossRef] [PubMed]
Cecconi, M.; Piovani, D.; Brunetta, E.; Aghemo, A.; Greco, M.; Ciccarelli, M.; Angelini, C.; Voza, A.; Omodei, P.; Vespa, E.; et al. Early predictors of clinical deterioration in a cohort of 239 patients hospitalized for COVID-19 infection in Lombardy, Italy. J. Clin. Med.2020, 9, 1548. [Google Scholar] [CrossRef] [PubMed]
Hanft, K. Lean Hospitals: Improving Quality, Patient Safety, and Employee Engagement, 2nd ed.; Productivity Press: Seattle, WA, USA, 2013; Volume 7, pp. 124–125. [Google Scholar]
Rotter, T.; Plishka, C.; Lawal, A.; Harrison, L.; Sari, N.; Goodridge, D.; Flynn, R.; Chan, J.; Fiander, M.; Poksinska, B.; et al. What is lean management in health care? Development of an operational definition for a cochrane systematic review. Eval. Health Prof.2019, 42, 366–390. [Google Scholar] [CrossRef] [PubMed]
Parkhi, S.S. Lean management practices in healthcare sector: A literature review. Benchmarking Int. J.2019, 26, 1275–1289. [Google Scholar] [CrossRef]
Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. Clinical characteristics of coronavirus disease 2019 in China. N. Engl. J. Med.2020, 382, 1708–1720. [Google Scholar] [CrossRef]
V’Kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol.2021, 19, 155–170. [Google Scholar] [CrossRef]
Kirtipal, N.; Bharadwaj, S.; Kang, S.G. From SARS to SARS-CoV-2, insights on structure, pathogenicity and immunity aspects of pandemic human coronaviruses. Infect. Genet. Evol.2020, 85, 104502. [Google Scholar] [CrossRef]
Livingston, E.; Bucher, K. Coronavirus disease 2019 (COVID-19) in Italy. JAMA2020, 323, 1335. [Google Scholar] [CrossRef]
Cucinotta, D.; Vanelli, M. WHO declares COVID-19 a pandemic. Acta Biomed.2020, 91, 157–160. [Google Scholar] [CrossRef] [PubMed]
World Health Organization. Clinical Management of Severe Acute Respiratory Infection (SARI) when COVID-19 Disease Is Suspected: Interim Guidance, 13 March 2020; World Health Organization: Geneva, Switzerland, 2020. [Google Scholar]
Percudani, M.; Corradin, M.; Moreno, M.; Indelicato, A.; Vita, A. Mental health services in lombardy during COVID-19 outbreak. Psychiatry Res.2020, 288, 112980. [Google Scholar] [CrossRef] [PubMed]
Odone, A.; Delmonte, D.; Scognamiglio, T.; Signorelli, C. COVID-19 deaths in Lombardy, Italy: Data in context. Lancet Public Health2020, 5, e310. [Google Scholar] [CrossRef]
Grasselli, G.; Zangrillo, A.; Zanella, A.; Antonelli, M.; Cabrini, L.; Castelli, A.; Cereda, D.; Coluccello, A.; Foti, G.; Fumagalli, R.; et al. Baseline characteristics and outcomes of 1591 patients infected with SARS-CoV-2 admitted to ICUs of the Lombardy region, Italy. JAMA2020, 323, 1574–1581. [Google Scholar] [CrossRef] [PubMed]
Masetti, C.; Generali, E.; Colapietro, F.; Voza, A.; Cecconi, M.; Messina, A.; Omodei, P.; Angelini, C.; Ciccarelli, M.; Badalamenti, S.; et al. High mortality in COVID-19 patients with mild respiratory disease. Eur. J. Clin. Investig.2020, 50, e13314. [Google Scholar] [CrossRef]
Zaim, S.; Chong, J.H.; Sankaranarayanan, V.; Harky, A. COVID-19 and multiorgan response. Curr. Probl. Cardiol.2020, 45, 100618. [Google Scholar] [CrossRef]
Noris, M.; Benigni, A.; Remuzzi, G. The case of complement activation in COVID-19 multiorgan impact. Kidney Int.2020, 98, 314–322. [Google Scholar] [CrossRef]
Mokhtari, T.; Hassani, F.; Ghaffari, N.; Ebrahimi, B.; Yarahmadi, A.; Hassanzadeh, G. COVID-19 and multiorgan failure: A narrative review on potential mechanisms. J. Mol. Histol.2020, 51, 613–628. [Google Scholar] [CrossRef]
Babapoor-Farrokhran, S.; Gill, D.; Walker, J.; Rasekhi, R.T.; Bozorgnia, B.; Amanullah, A. Myocardial injury and COVID-19: Possible mechanisms. Life Sci.2020, 253, 117723. [Google Scholar] [CrossRef]
Ronco, C.; Reis, T.; Husain-Syed, F. Management of acute kidney injury in patients with COVID-19. Lancet Respir. Med.2020, 8, 738–742. [Google Scholar] [CrossRef]
Jafari-Oori, M.; Fiorentino, M.; Castellano, G.; Ebadi, A.; Rahimi-Bashar, F.; Guest, P.C.; Vahedian-Azimi, A.; Sahebkar, A. Acute kidney injury and COVID-19: A scoping review and meta-analysis. Adv. Exp. Med. Biol.2021, 1321, 309–324. [Google Scholar] [CrossRef] [PubMed]
Lee, I.C.; Huo, T.I.; Huang, Y.H. Gastrointestinal and liver manifestations in patients with COVID-19. J. Chin. Med. Assoc.2020, 83, 521–523. [Google Scholar] [CrossRef]
Jothimani, D.; Venugopal, R.; Abedin, M.F.; Kaliamoorthy, I.; Rela, M. COVID-19 and the liver. J. Hepatol.2020, 73, 1231–1240. [Google Scholar] [CrossRef]
Baig, A.M. Neurological manifestations in COVID-19 caused by SARS-CoV-2. CNS Neurosci. Ther.2020, 26, 499–501. [Google Scholar] [CrossRef]
Whittaker, A.; Anson, M.; Harky, A. Neurological manifestations of COVID-19: A systematic review and current update. Acta Neurol. Scand.2020, 142, 14–22. [Google Scholar] [CrossRef] [PubMed]
Aghemo, A.; Piovani, D.; Parigi, T.L.; Brunetta, E.; Pugliese, N.; Vespa, E.; Omodei, P.D.; Preatoni, P.; Lleo, A.; Repici, A.; et al. COVID-19 digestive system involvement and clinical outcomes in a large academic hospital in Milan, Italy. Clin. Gastroenterol. Hepatol.2020, 18, 2366–2368.e2363. [Google Scholar] [CrossRef]
Ferrante, G.; Fazzari, F.; Cozzi, O.; Maurina, M.; Bragato, R.; D’Orazio, F.; Torrisi, C.; Lanza, E.; Indolfi, E.; Donghi, V.; et al. Risk factors for myocardial injury and death in patients with COVID-19: Insights from a cohort study with chest computed tomography. Cardiovasc. Res.2020, 116, 2239–2246. [Google Scholar] [CrossRef]
Cocco, A.; Amami, P.; Desai, A.; Voza, A.; Ferreli, F.; Albanese, A. Neurological features in SARS-CoV-2-infected patients with smell and taste disorder. J. Neurol.2021, 268, 1570–1572. [Google Scholar] [CrossRef]
Desai, A.; Voza, G.; Paiardi, S.; Teofilo, F.I.; Caltagirone, G.; Pons, M.R.; Aloise, M.; Kogan, M.; Tommasini, T.; Savevski, V.; et al. The role of anti-hypertensive treatment, comorbidities and early introduction of LMWH in the setting of COVID-19: A retrospective, observational study in Northern Italy. Int. J. Cardiol.2021, 324, 249–254. [Google Scholar] [CrossRef] [PubMed]
Luzzi, S.; Giotta Lucifero, A.; Marasco, S.; Del Maestro, M.; Bellantoni, G.; Gragnaniello, C. Targeting of renin-angiotensin system in COVID-19 patients affected by stroke: Emerging concerns about detrimental vs. benefit effect. Interdiscip. Neurosurg.2020, 22, 100822. [Google Scholar] [CrossRef] [PubMed]
Desai, A.; Caltagirone, G.; Sari, S.; Pocaterra, D.; Kogan, M.; Azzolini, E.; Savevski, V.; Martinelli-Boneschi, F.; Voza, A. The Use of Antiviral Agents against SARS-CoV-2: Ineffective or Time and Age Dependent Result? A Retrospective, Observational Study among COVID-19 Older Adults. J. Clin. Med.2021, 10, 686. [Google Scholar] [CrossRef]
Desai, A.; Santonocito, O.G.; Caltagirone, G.; Kogan, M.; Ghetti, F.; Donadoni, I.; Porro, F.; Savevski, V.; Poretti, D.; Ciccarelli, M.; et al. Effectiveness of streptococcus pneumoniae urinary antigen testing in decreasing mortality of COVID-19 co-infected patients: A clinical investigation. Medicina2020, 56, 572. [Google Scholar] [CrossRef]
Kilaru, A.S.; Lee, K.; Snider, C.K.; Meisel, Z.F.; Asch, D.A.; Mitra, N.; Delgado, M.K. Return hospital admissions among 1419 COVID-19 patients discharged from five U.S. emergency departments. Acad. Emerg. Med.2020, 27, 1039–1042. [Google Scholar] [CrossRef] [PubMed]
Gok, A.F.K.; Eryılmaz, M.; Ozmen, M.M.; Alimoglu, O.; Ertekin, C.; Kurtoglu, M.H. Recommendations for trauma and emergency general surgery practice during COVID-19 pandemic. Ulus. Travma Acil Cerrahi Derg.2020, 26, 335–342. [Google Scholar] [CrossRef]
De Freitas, L.; Goodacre, S.; O’Hara, R.; Thokala, P.; Hariharan, S. Interventions to improve patient flow in emergency departments: An umbrella review. Emerg. Med. J.2018, 35, 626–637. [Google Scholar] [CrossRef] [PubMed]
Giamberardino, M.A.; Affaitati, G.; Costantini, R.; Guglielmetti, M.; Martelletti, P. Acute headache management in emergency department. A narrative review. Intern. Emerg. Med.2020, 15, 109–117. [Google Scholar] [CrossRef] [PubMed]
Cardona, M.; Lewis, E.T.; Turner, R.M.; Alkhouri, H.; Asha, S.; Mackenzie, J.; Perkins, M.; Suri, S.; Holdgate, A.; Winoto, L.; et al. Efficacy of a tool to predict short-term mortality in older people presenting at emergency departments: Protocol for a multi-centre cohort study. Arch. Gerontol. Geriatr.2018, 76, 169–174. [Google Scholar] [CrossRef]
Savioli, G.; Ceresa, I.F.; Luzzi, S.; Gragnaniello, C.; Giotta Lucifero, A.; Del Maestro, M.; Marasco, S.; Manzoni, F.; Ciceri, L.; Gelfi, E.; et al. Rates of intracranial hemorrhage in mild head trauma patients presenting to emergency department and their management: A comparison of direct oral anticoagulant drugs with vitamin k antagonists. Medicina2020, 56, 308. [Google Scholar] [CrossRef] [PubMed]
Savioli, G.; Ceresa, I.F.; Ciceri, L.; Sciutti, F.; Belliato, M.; Iotti, G.A.; Luzzi, S.; Del Maestro, M.; Mezzini, G.; Lafe, E.; et al. Mild head trauma in elderly patients: Experience of an emergency department. Heliyon2020, 6, e04226. [Google Scholar] [CrossRef] [PubMed]
Savioli, G.; Ceresa, I.F.; Macedonio, S.; Gerosa, S.; Belliato, M.; Iotti, G.A.; Luzzi, S.; Del Maestro, M.; Mezzini, G.; Giotta Lucifero, A.; et al. Trauma coagulopathy and its outcomes. Medicina2020, 56, 205. [Google Scholar] [CrossRef]
Khanna, S.; Sier, D.; Boyle, J.; Zeitz, K. Discharge timeliness and its impact on hospital crowding and emergency department flow performance. Emerg. Med. Australas2016, 28, 164–170. [Google Scholar] [CrossRef]
Costa, L.B.; Filho, M.G.; Rentes, A.F.; Bertani, T.M.; Mardegan, R. Lean healthcare in developing countries: Evidence from Brazilian hospitals. Int. J. Health Plann. Manag.2017, 32, e99–e120. [Google Scholar] [CrossRef]
Mohammadian, M.; Babaei, M.; Amin Jarrahi, M.; Anjomrouz, E. Scheduling nurse shifts using goal programming based on nurse preferences: A case study in an emergency department. Int. J. Eng.2019, 32, 954–963. [Google Scholar] [CrossRef]
Pitts, S.R.; Niska, R.W.; Xu, J.; Burt, C.W. National hospital ambulatory medical care survey: 2006 emergency department summary. In National Health Statistics Reports; U.S. Department of Health & Human Services: Washington, WA, USA, 2008; pp. 1–38. [Google Scholar]
Klein, D.; Khan, V. Utilizing six sigma lean strategies to expedite emergency department CT scan throughput in a tertiary care facility. J. Am. Coll. Radiol.2017, 14, 78–81. [Google Scholar] [CrossRef]
Khlie, K.; Abouabdellah, A. Identification of the patient requirements using lean six sigma and data mining. Int. J. Eng.2017, 30, 691–699. [Google Scholar]
Sánchez, M.; Suárez, M.; Asenjo, M.; Bragulat, E. Improvement of emergency department patient flow using lean thinking. Int. J. Qual. Health Care2018, 30, 250–256. [Google Scholar] [CrossRef] [PubMed]
Dickson, E.W.; Singh, S.; Cheung, D.S.; Wyatt, C.C.; Nugent, A.S. Application of lean manufacturing techniques in the Emergency Department. J. Emerg. Med.2009, 37, 177–182. [Google Scholar] [CrossRef] [PubMed]
Holden, R.J. Lean Thinking in emergency departments: A critical review. Ann. Emerg. Med.2011, 57, 265–278. [Google Scholar] [CrossRef] [PubMed]
Improta, G.; Romano, M.; Di Cicco, M.V.; Ferraro, A.; Borrelli, A.; Verdoliva, C.; Triassi, M.; Cesarelli, M. Lean thinking to improve emergency department throughput at AORN Cardarelli hospital. BMC Health Serv. Res.2018, 18, 914. [Google Scholar] [CrossRef] [PubMed]
Aminjarahi, M.; Abdoli, M.; Fadaee, Y.; Kohan, F.; Shokouhyar, S. The prioritization of lean techniques in Emergency Departments using VIKOR and SAW approaches. Ethiop. J. Health Sci.2021, 31, 283–292. [Google Scholar] [CrossRef] [PubMed]
Dr. Carlos Alberto Díaz. Profesor Titular UNIVERSIDAD ISALUD. Buenos Aires. Argentina.
Omicron Tiene otras características clínicas, se requiere otro modelo prestacional, que es: aumentar la producción de testeos, triplicar los centros de febriles, vacunar nuevamente en los vacunatorios oficiales, colocar incentivos para que la gente se vacune, aislar a los contactos, seguir a los pacientes telefónicamente, limitar días de aislamiento, seguir brindando servicios asistenciales para no demorar la atención de otras patologías, desarrollar nuevas vacunas más potentes y contra las nuevas cepas, incentivar la realización de autotest, para limitar la concurrencia y mejorar acceso a los antivirales para limitar el periodo de estado y las internaciones.
Los próximos días serán record de contagios.
Hoy en España 100 mil casos nuevos, Argentina 33 mil, pronto llegaremos a esas cifras con incidencia record.1900 casos cada 100 mil habitantes.Algunos encuentros sociales hay que limitar los eventos recreativos masivos en locales cerrados en los próximos quince días.
Mañana 29 de diciembre superaremos la cantidad de casos diarios récord en Argentina. Es otra enfermedad, por el covid 19, es diferente epidemiológicamente y clínicamente. Exige otro modelo prestacional asistencial, con más sitios de diagnóstico y triage con SCORE NEWS 2 para acelerar procesos de estadificación y aislamiento. Estas respuestas son difíciles porque son de escalada rápida y desactivación en cuatro semanas, es complejo para los sistemas de salud que no tienen oferta redundante desplegar una mayor oferta. Además el personal está defraudado, ya no quiere más dar algo más y arriesgarse. Están trabajando a reglamento. Los dirigentes no lo pueden decir, porque estarían reconociendo su ineptitud, su cinismo, lo mentirosos que son, y que la salud no les importa nada, nada, nunca fue para las gestiones políticas una prioridad, y menos que en los cargos hay especialistas sin peso político propio, como tenía Ginés, que podía levantar la voz, e igual se lo llevaron puesto, porque no pudieron reconocer que era algo que ellos mismos sabían, ellos lo conocían. No hay que olvidar que los testeados fueron personas que viajaban a una misión a México, por el tema del acuerdo de producción de la vacuna de adenovirus de Oxford Astra Zeneca. No olvidemos. Las sociedades que no tienen memoria, están destinadas al fracaso. Las sociedades que no se recuerda lo que le hicieron sus gobernantes y los reeligen porque le prometieron que volvían diferentes, solo grandes hombres pueden hacer eso, y no es con lo que estamos tratando. Son opositores y oficialistas iguales. Son lo mismo y defienden sus privilegios de burguesía. La diferencia con Brasil que esta Burguesía es política no productiva, y no generan riqueza.
Todos los días si no hacemos nada distinto todos los números exponencialmente crecerán semana tras semana, para llegar a los cienmil casos diarios en pocas jornadas, sin asustarnos, el mayor problema estará en cómo haremos los testeos, cuantos lugares, con que metodología, creo que los pacientes se tienen que testear sin que los vea un médico o los examine, pueda hacersetest de antígenos en estaciones, para que se realice por etapas en una producción industrial, hace falta hacer más testeos, si, en nuestra realidad hoy tenemos el pico más alto de toda la pandemia por un total de 47%, por lo tanto no son personas que vienen a testearse por las dudas.
Hoy para llevar al número de positividad que requiere la OMS, tendríamos que habernos acercado a 300.000 testeos, si hay que triplicar la capacidad, las Unidades febriles están al tope y no los 116.000 que se hicieron, 33.900 casos positivos. Concentrar el esfuerzo en colocar al personal para testear.
Simultáneamente hay que vacunar al doble del ritmo actual de lo que venimos haciendo hoy. Concientizar, poner incentivos, citar, dar confianza a la población. Ampliar los lugares de vacunación, llevarlos a los lugares habituales con los vacunadores oficiales. Astra Zeneca, puede guardarse en una heladera convencional por ello se puede expandir mucho los sitios de vacunación.
Cambiar las medidas de aislamiento y duración de contacto estrecho:
Aislamiento personal de salud, como contacto estrecho. El problema además que por cada caso hay una decena por lo menos de contactos estrechos, entre familias y empleados, por lo tanto no se puede seguir con estos esquemas de aislamiento. hay que mejorar las medidas de aislamiento.
Los contactos estrechos son tantos que no se puede seguir por este camino. Nuevamente hay que tomar medidas diferentes.
Debemos hacer cosas distintas. Los dispositivos asistenciales deben modificarse, aumentar la teleconsulta y seguimiento, como así también acceso a las guardias de los casos graves, Estamos en un nivel 3 de riesgo de infección para el CDC. Ocho mil cancelaciones de aviones. No tendremos personal de salud para atender. No vamos a poder identificar los casos reales.
En Canadá, en Quebec están dejando permitir que el personal de salud pueda trabajar contagiado. si como se escucha. Hace un año teníamos 7.216 casos, hoy 33.900 el más alto fue de 35.017 en un día, mañana estaremos superando el máximo, con ocho mil camas ocupadas de terapia intensiva, la buena noticia tenemos 34% de camas de terapia intensiva. Hace quince días teníamos 3.512 casos. Quebec permite que algunos trabajadores de la salud infectados permanezcan en el trabajo a medida que aumentan las ausencias relacionadas con COVID. No me parece correcto, solo lo cito, para que observemos que pasa en el resto del mundo. El ministro de Salud de Quebec, Christian Dubé, que se mostró el 16 de diciembre en Montreal, anunció el martes que algunos trabajadores de la salud en la provincia que dieron positivo por COVID-19 o estuvieron en contacto cercano con un caso confirmado pueden permanecer en el trabajo para proteger capacidad hospitalaria. (Paul Chiasson / The Canadian Press).
El ministro de salud de Quebec anunció el martes que algunos trabajadores de la salud en la provincia que dieron positivo por COVID-19 o estuvieron en contacto cercano con un caso confirmado pueden permanecer en el trabajo para proteger la capacidad del hospital.
La provincia planea extender la política a todos los trabajadores esenciales en los próximos días y semanas. El ministro de Salud, Christian Dubé, dijo que la decisión se tomó como una forma de «gestión de riesgos», ya que el sistema de atención médica experimenta miles de ausencias relacionadas con COVID.
Dubé dijo que el gobierno ha estado en conversaciones con los sindicatos durante varios días sobre el cambio de política. Dijo que llegaron a la conclusión de que era la única forma de prevenir «daños permanentes» para las personas que necesitan atención médica y que las decisiones se tomarán caso por caso.
«La realidad es que tenemos cada vez más enfermos y cada vez menos personal de enfermería», dijo. «Esto es lo que tenemos que hacer si queremos que nuestra sociedad continúe funcionando con seguridad».
El ministro dijo que había alrededor de 7.000 trabajadores de la salud fuera del trabajo el lunes debido al COVID-19, y se espera que ese número aumente a 10.000 en los próximos días. Esas ausencias temporales se suman a las miles de enfermeras que dejaron el sistema público durante la pandemia por el precio que estaba cobrando.
«Si tuviéramos la opción, no lo haríamos, pero nuestra situación es urgente y crítica en el corto plazo», dijo Dubé. «Esta es la mejor alternativa a no brindar atención».
Sería notar que las infecciones por Ómicron causan pocos síntomas leves en la mayoría o en todos los que se infectan. Incluso con altas tasas de transmisión y una gran cantidad de casos entre vacunados, una variante que cause poco más que resfríos o algunos días de fatiga podría ser bienvenida como el comienzo de la endemicidad, un estado en el que el virus permanece entre nosotros indefinidamente, que podría marcar el comienzo del fin de la pandemia.
Nuevas vacunas:
Pfizer y BioNTech empezaron a desarrollar una versión específica para el ómicron de sus vacunas el 25 de noviembre y afirmaron que los primeros lotes podrían estar listos para su entrega en 100 días, a la espera de la aprobación reglamentaria. Moderna también está trabajando en una versión actualizada contra la nueva variante y dijo que podría terminar las pruebas y estar lista para presentarla a los reguladores en marzo de 2022.
Sin embargo, en la sesión informativa del Centro de Medios de Comunicación Científica de Alemania en relación con la vacuna de Oxford-AstraZeneca, Teresa Lambe, investigadora principal del Instituto Jenner de la Universidad de Oxford, dijo: «No sabemos todavía si necesitamos una nueva vacuna. Es muy, muy probable que veamos un descenso de los anticuerpos neutralizantes, [pero] todavía no hemos encontrado una variante en la que hayamos visto un impacto en la protección contra la hospitalización y la muerte. Desgraciadamente, tenemos que ser un poco pacientes para que salgan esos datos. Nosotros, como otros fabricantes de vacunas, podemos ir rápido. Ya hemos fabricado una vacuna diferente, AZD2816, para la variante beta, así que tenemos los procesos y la fuerza de voluntad para ir rápido si lo necesitamos«.
Dado lo que sabemos actualmente sobre COVID-19 y la variante Omicron, los CDC están acortando el tiempo recomendado para el aislamiento de 10 días para las personas con COVID-19 a 5 días, si son asintomáticos, seguidos de 5 días de uso de una máscara cuando están cerca de otros.
El cambio está motivado por la ciencia que demuestra que la mayoría de la transmisión del SARS-CoV-2 ocurre temprano en el curso de la enfermedad, generalmente en los 1-2 días anteriores al inicio de los síntomas y los 2-3 días después. Por lo tanto, las personas que dan positivo deben aislarse durante 5 días y, si son asintomáticas en ese momento, pueden dejar el aislamiento si pueden continuar enmascarando durante 5 días para minimizar el riesgo de infectar a otros.
Para las personas que no están vacunadas o que están a más de seis meses de su segunda dosis de ARNm (o más de 2 meses después de la vacuna J&J) y aún no se han reforzado, los CDC ahora recomiendan la cuarentena durante 5 días seguidos del uso estricto de la máscara durante 5 días adicionales.
Alternativamente, si una cuarentena de 5 días no es factible, es imperativo que una persona expuesta use una máscara bien ajustada en todo momento cuando esté cerca de otras personas durante 10 días después de la exposición.
Las personas que han recibido su vacuna de refuerzo no necesitan ponerse en cuarentena después de una exposición, pero deben usar una máscara durante 10 días después de la exposición.
Para todos los expuestos, la mejor práctica también incluiría una prueba de SARS-CoV-2 al día 5 después de la exposición.
Si se presentan síntomas, las personas deben ponerse en cuarentena inmediatamente hasta que una prueba negativa confirme que los síntomas no son atribuibles a COVID-19.
El aislamiento se relaciona con el comportamiento después de una infección confirmada. El aislamiento durante 5 días seguido de usar una máscara bien ajustada minimizará el riesgo de propagar el virus a otras personas.
La cuarentena se refiere al tiempo posterior a la exposición al virus o al contacto cercano con alguien que se sabe que tiene COVID-19.
Ambas actualizaciones se producen a medida que la variante Omicron continúaextendiéndose por todo Estados Unidos y refleja la ciencia actual sobre cuándo y durante cuánto tiempo una persona es máximamente infecciosa.
Los datos de Sudáfrica y el Reino Unido demuestran que la efectividad de la vacuna contra la infección para dos dosis de una vacuna de ARNm es de aproximadamente el 35%. Una dosis de refuerzo de la vacuna COVID-19 restaura la efectividad de la vacuna contra la infección al 75%.
La vacunación contra el COVID-19 disminuye el riesgo de enfermedad grave, hospitalización y muerte por COVID-19. Los CDC recomiendan encarecidamente la vacunación contra el COVID-19 para todas las personas mayores de 5 años y los refuerzos para todas las personas mayores de 16 años. La vacunación es la mejor manera de protegerse y reducir el impacto de COVID-19 en nuestras comunidades.
Lo siguiente es atribuible a la Directora de los CDC, la Dra. Rochelle Walensky:
«La variante Omicron se está extendiendo rápidamente y tiene el potencial de impactar todas las facetas de nuestra sociedad. Las recomendaciones actualizadas de los CDC para el aislamiento y la cuarentena equilibran lo que sabemos sobre la propagación del virus y la protección proporcionada por la vacunación y las dosis de refuerzo. Estas actualizaciones garantizan que las personas puedan continuar su vida diaria de manera segura. La prevención es nuestra mejor opción: vacunarse, impulsarse, usar una máscara en entornos públicos en interiores en áreas de transmisión comunitaria sustancial y alta, y hacerse una prueba antes de reunirse».Si da positivo por COVID-19 (Aislar)
Todos, independientemente del estado de vacunación.
Quédese en casa durante 5 días.
Si no tiene síntomas o sus síntomas se están resolviendo después de 5 días, puede salir de su casa.
Continúe usando una máscara alrededor de otras personas durante 5 días adicionales.
Si tiene fiebre, continúe quedándose en casa hasta que la fiebre se resuelva.Si estuvo expuesto a alguien con COVID-19 (cuarentena)
Quédese en casa durante 5 días. Después de eso, continúe usando una máscara alrededor de otros durante 5 días adicionales.
Si no puede ponerse en cuarentena, debe usar una máscara durante 10 días.
Prueba el día 5 si es posible.
Si desarrolla síntomas, hágase una prueba y quédese en casa
Estamos en la tercer ola Donde cabalgan dos cepas Ómicron y Delta, por suerte estamos frente a esta ola con vacunados en el 70 por ciento con dos dosis, pero pocos con tres dosis, 9,5%, lo que hoy se considera oficialmente vacunación completa, la ómicron y la delta tiene una tasa de reinfección más alto. Pero el relajamiento supera la disciplina social y las fiestas masivas impresionantes el fin de semana.Una parte de esta sociedad tiene una baja percepción del riesgo y de la interacción social.
Si la gente se sigue juntando esto va a explotar por los aires. Hay que limitar los números de contagiados. Seguir impulsando las medidas no farmacológicas. Hay que recordar que el barbijo protege, bajan muchísimo la incidencia del contagio. Debemos empoderar a la sociedad. Todas las vacunas funcionan.
Debemos acelerar la vacunación, implementar más sitios de vacunación, liberarla y aumentar los testeos.
Una campaña de comunicación es muy deficitaria oficial, porque no se muestra todo lo bueno que tiene estar vacunado.
Nos quedamos en modo electoral, que todo había terminado. Hoy hay muy pocos internados, casi ninguno de los graves de hoy internados en terapia intensiva, tiene la vacunación completa, esto sería un incentivo poderosísimo para que se vacunen.
Estas subidas de casos no se traducen en más internaciones o complicaciones. Por ello, pensamos que puede ser el fin de la pandemia. Revisar los protocolos de aislamiento siguiendo al CDC.
Los servicios de testeo están con varias cuadras de cola, se duplicaron la cantidad de casos y a nadie le importa. La gente descree de los contagios. Pero cuando se siente mal recurre a los servicios de salud y a sus trabajadores para que los asistan, cuando no los reconoce como se debe. Cuando sus representantes los engañaron, y ahora no están dispuestos a dar la vida por unos líderes traidores.
Hay que autorizar los auto-testeos especialmente al personal de salud, a los servidores públicos, en las escuelas, en los jardines, en las colonias de vacaciones.
El gobierno trasunta un falso optimismo, porque ganó perdiendo en las urnas, porque la oposición no sabe que hacer con el triunfo, decretó por DNU que la epidemia ya paso, la gente salió a divertirse en forma inusitada, que se termina el planeta, y aumentan con esas fiestas masivas exponencialmente los contagios.
La gente se violenta porque lo hacen esperar, porque quiere testearse para seguir repicando en las fiestas.
Propio de una ciudadanía que no ven que la ley sea importante, por ello la ley es solamente un elemento referencial en argentina, no algo que sirva para poner límites a conductas inadecuadas de la vida en sociedad, ya que los que triunfan son transgresores. Se impone la doble moral de los maradonianos de siempre, “las alegrías que nos dio” “con él salimos campeones” o los Camporistas “tuvimos una casa y no sé cuántos justificativos”, para decir elijo por esto que es lo menos malo que algo me dio.
Se transfieren constantemente responsabilidades, no se comienza por la responsabilidad individual personal, de quererse como persona y a su familia, no importa contagiar a inmunocomprometidos, que hace dos años que están prácticamente recluidos.
La ley en argentina es una tentación para ser transgredida, parece que no está promulgada para todos, hay que transgredirla y vanagloriarse de ello de esas conductas arriesgadas.
Hacen actos multitudinarios para frenar a los albertistas que no se crean que ya pueden gobernar sin la señora porque todavía no completaron su trabajo de que vuelva impoluta a ser candidata, porque sacándose definitivamente el cuero de oveja, ahora quiere volver con todos los honores y santa eva, ya no alcanza con salvar a los hijos, ahora nos quiere sojuzgar por el lado que ella desea porque considera que es la poseedora de la verdad, de la manera de hacer las cosas, ¿porque le tienen tanto miedo quienes gobiernan?, que rompen sus propios pares de origen, que es lo que los lleva a temerle, porque si no tuviéramos algunos que ella señala correctamente como sus enemigos, en el periodismo y en la corte suprema estaríamos liquidados, los opositores son una sarta de incompetentes agrupados que volvieron a sus andanzas no respetando el mandato que les había dado el gobierno, no aumenten los impuestos y a ala vuelta del primer triunfo firmar un pacto fiscal para aumentar los impuestos. Reúnen dos criterios mayores son malnacidos y temerosos. Además de hipócritas y cínicos. Son tantas las virtudes. Es una casta de acomodaticios. Que preservan solamente sus privilegios de burgueses políticos, que discuten como déspotas ilustrados, si pueden ser reelegidos o no. La gente quiere trabajo, seguridad, que no haya inflación, que puedan darle un futuro a sus hijos.
Es inconcebible, sorprendente, que encima quieren porque sacaron dos votos más que en anteriores elecciones y gobiernan una pequeña provincia, querer mandar en todo el país. Pelearse antes de lograr el objetivo, propios del partido radical. Boicotearse. Ser enemigos de su triunfo. Suicidarse. Darle lugar a los que los ciudadanos no quieren.
Los penalizan por tener más de 0,50 de alcoholemia manejando, al final no nos dejar festejar la navidad dicen los penalizados.
Las personas no quieren inflación, sino quieren trabajar dignamente, tener un futuro para sus hijos, tener seguridad, un sistema educativo que impulse e incentive el ascenso social, por el esfuerzo, no con los planes o por ser camporistas.
La oposición vergonzante, no sabe que hacer con ese triunfo electoral, y nos entregan a este falso gobierno populista, porque no hace nada por los pobres y para redistribuir la riqueza más allá de los planes de subsidio. La gente los mira, les pide que estén unidos y que sigan mirando los problemas de la gente y no los de la política.
Reference Pricing in Germany: Implications for U.S. Pharmaceutical Purchasing
Para realizar este posteo se analizó el comportamiento de esta metodología de negociación conocida como precios de referencia de los medicamentos, para el estímulo de la competencia, y disminuir la dispersión del precio. Aunque deberíamos colocar incentivos para que los que están muy abajo, no practiquen el efecto murciélago y descansen colgando del techo en los precios, como comportamiento oportunista. Instaló este tema porque puede ser impulsado por el Ministerio de Salud en Argentina. Tenemos como antecedente hace más de dos décadas un sistema en la Obra social Provincial que fue exitoso. Como también el que implementé en una Mutual. El comportamiento inicial es que si los líderes en el segmento están muy por encima moderan los incrementos de precios. Lo difícil en este contexto inflacionario es sostener una modalidad cuya evaluación inicial lleva un año, y podría llevar a acuerdos ex ante, entre las farmacéuticas y asegurarse una base anticipada de rentabilidad. Es una buena herramienta, el secreto estará en una buena posible implementación, y empezaría por los medicamentos que son más utilizados, un número de 1200 productos farmacéuticos, para empezar, luego ir evolucionando hacia el resto. Además en argentina no tenemos una agencia de evaluación de tecnologías y esto haría una batalla despiadada de lobbies que no se quién estaría dispuesto a librar.
«Se considera precio de referencia la cuantía máxima que se financiará con cargo a fondos de la Seguridad Social o fondos afectos a la sanidad, de las presentaciones de especialidades farmacéuticas incluidas en cada uno de los conjuntos homogéneos que se determinen.»
El precio de cada conjunto debe calcularse atendiendo al coste diario del tratamiento más barato del conjunto de referencia cuyo precio quiere calcularse. Es decir, en este sistema se clasifican los medicamentos principalmente agrupándolos por principio activo y vía de administración, de cada conjunto se determina qué presentación es la que tiene un coste/diario menor y éste determina el precio de referencia del conjunto. En base al precio de referencia del conjunto, se calcula el precio de referencia de cada una de las presentaciones.
De acuerdo con la normativa, el precio de referencia de cada presentación debe calcularse multiplicando el precio de referencia del conjunto por el número de dosis diarias definidas (DDD) contenidas en cada presentación.
Los precios de referencia, son un componente del diseño del seguro de salud, un comprador de atención médica establece un pago máximo que contribuirá a cubrir el precio de un medicamento. Se utiliza cuando hay una amplia variación en los precios de productos terapéuticamente similares. El límite de pago se establece en el punto mínimo, mediano u otro punto a lo largo del rango de precios de los medicamentos dentro de una clase terapéutica. Si el médico de un paciente prescribe un medicamento con un precio igual o inferior al límite de referencia, el paciente paga solo un copago modesto. Si se selecciona una opción más cara, él o ella paga el copago más la diferencia total entre el límite de referencia y el precio del producto elegido.
Los precios de referencia ofrecen varias ventajas sobre los diseños de seguros más utilizados en los Estados Unidos, como los deducibles anuales y el coseguro, que exponen a los consumidores a obligaciones financieras sin proporcionar una opción asequible u orientación sobre cómo seleccionar productos que ofrezcan el mejor valor. Hasta la fecha, sin embargo, los precios de referencia sólo han sido aplicados por un número limitado de compradores y sólo a las clases de medicamentos que presentan múltiples alternativas genéricas o terapéuticamente equivalentes. Para estas clases terapéuticas, se puede suponer razonablemente que todos los productos funcionan de manera similar.
Los compradores pueden limitar sus pagos al nivel cobrado por los productos más baratos en cada clase y los pacientes que deseen una opción de mayor precio razonablemente pueden ser obligados a pagar la diferencia ellos mismos. A los pacientes con necesidades clínicas identificadas por el médico para opciones de mayor precio se les puede otorgar una excepción.
En sus esfuerzos por mejorar la eficacia y la eficiencia de las compras farmacéuticas, Estados Unidos puede aprender de Alemania, que gestiona los medicamentos tradicionales utilizando precios de referencia y los nuevos medicamentos utilizando precios de eficacia comparativa.
Alemania ha desarrollado métodos basados en la evidencia para evaluar el beneficio clínico de los nuevos productos, establecer pagos basados en referencia para los medicamentos que no ofrecen beneficios incrementales sobre los productos existentes y negociar nuevos precios para los medicamentos que sí ofrecen beneficios incrementales.1
Este enfoque goza de una considerable legitimidad social como mecanismo para garantizar el acceso de los pacientes al tiempo que modera los gastos de los pagadores.
El sistema de atención médica en Alemania se asemeja al de los Estados Unidos en varios aspectos importantes, pero difiere en otros. Ambos cuentan con múltiples aseguradoras no gubernamentales en lugar de un solo pagador gubernamental, favorecen la negociación sobre la regulación para determinar los precios, disfrutan de gastos decrecientes para muchos medicamentos tradicionales no especializados, pero enfrentan gastos crecientes para nuevos productos especializados, y están integrados en una cultura que valora el acceso de los pacientes incluso a los tratamientos más caros. Sin embargo, en Alemania, la evaluación clínica de cada nuevo medicamento está centralizada y la negociación de los precios de los medicamentos se realiza colectivamente por la organización paraguas de las aseguradoras de salud, en lugar de por cada aseguradora individualmente. Este resumen de la edición describe la estructura de la evaluación y fijación de precios de medicamentos en Alemania y su posible aplicabilidad al mercado estadounidense.2
El marco institucional de la fijación de precios farmacéuticos en Alemania
En Alemania, los precios de referencia se encuentran dentro de un sistema institucional que cuenta con asociaciones de aseguradoras, médicos y otras partes interesadas reguladas y responsables públicamente. La legislación y la jurisprudencia establecen las normas que rigen las interacciones entre estas entidades, y el Ministerio de Salud supervisa y apoya continuamente sus procesos. Pero el gobierno no evalúa directamente el beneficio clínico comparativo de los nuevos medicamentos ni negocia sus precios. En este sentido, se parece más al marco de los Estados Unidos que a otros sistemas europeos donde el trabajo pesado en el control de costos farmacéuticos es realizado directamente por los pagadores gubernamentales.
El marco institucional alemán difiere de su homólogo estadounidense en aspectos importantes. La organización que evalúa el rendimiento clínico comparativo de los nuevos medicamentos, el Comité Conjunto Federal (GBA), está formado por representantes de las organizaciones nacionales de seguros, médicos y hospitales. Las organizaciones de defensa de los pacientes tienen asientos sin derecho a voto en la junta. El GBA, a su vez, delega la evaluación clínica de nuevos medicamentos a una entidad privada pero públicamente responsable, el Instituto para la Calidad y Eficiencia en la Atención Médica (IQWiG). IQWiG basa sus evaluaciones en: expedientes presentados por los fabricantes, que incluyen una revisión sistemática del beneficio incremental del medicamento; los ensayos clínicos para la autorización inicial de comercialización por parte de la Agencia Europea de Medicamentos, así como otros ensayos clínicos; informes de organismos de evaluación de la tecnología de otros países; y otras pruebas disponibles. GBA luego hace su evaluación oficial de la contribución de cada medicamento basada en el estudio IQWiG, más aportes de los fabricantes y testimonios de seguimiento en reuniones públicas.
Las evaluaciones de GBA son utilizadas por la organización paraguas de las Cajas de Enfermedad, la GKV-SV. El GKV-SV trabaja dentro de un marco legal y regulatorio que le asigna derechos y responsabilidades especiales, e interpreta su papel como la negociación de los mejores precios desde el punto de vista del sistema de salud, y no simplemente el de sus aseguradoras constituyentes.
Evaluación de la eficacia comparativa
En el sistema farmacéutico alemán, los nuevos medicamentos se evalúan y fijan un precio en relación con los tratamientos existentes para las mismas afecciones. Los medicamentos que ofrecen beneficios clínicos adicionales reciben precios más altos; los precios de referencia se aplican a los nuevos medicamentos con un rendimiento clínico similar al de los productos que ya están en el mercado. Los precios de efectividad comparativa se aplican a los nuevos productos que funcionan mejor que sus comparadores.
Todos los medicamentos autorizados para el acceso al mercado por la Agencia Europea de Medicamentos (EMA) están disponibles inmediatamente después del lanzamiento para que los médicos los prescriban y los pacientes los usen. El fabricante establece unilateralmente el precio del nuevo medicamento en el momento del lanzamiento y se le reembolsa en su totalidad a ese precio durante el primer año del medicamento. Durante este primer año, se lleva a cabo una evaluación de la seguridad y eficacia clínica comparativa del medicamento por parte del Comité Conjunto Federal (GBA), una entidad autónoma pero públicamente responsable que representa a asociaciones de aseguradoras no gubernamentales (también conocidas como «Fondos de Enfermedad»), médicos y hospitales.
El GBA toma varias decisiones importantes con respecto a la evaluación del beneficio incremental de cada medicamento, con aportes del Instituto para la Calidad y Eficiencia en la Atención Médica (IQWiG), el fabricante farmacéutico, las asociaciones médicas relevantes, las organizaciones de defensa del paciente y otras entidades interesadas. En primer lugar, y a menudo lo más importante, GBA decide qué medicamento se utilizará como comparador con el que se evaluará el nuevo producto; un fármaco que trata múltiples indicaciones puede tener múltiples comparadores. Si se descubre que el nuevo medicamento ofrece beneficios incrementales, su precio se negociará al alza con respecto al precio del comparador, por lo que el fabricante tiene interés en que el GBA seleccione un comparador de alto precio. Sin embargo, si GBA elige como comparador un medicamento con alto precio pero también alta eficacia, el nuevo medicamento se enfrenta a un desafío más difícil para demostrar un beneficio incremental. Un hallazgo de ningún beneficio incremental lleva a que el medicamento se asigne a una clase terapéutica sujeta a precios de referencia. Todos los productos se reembolsan a un nivel basado en los precios más bajos cobrados dentro de la clase, si pertenece a una clase terapéutica para la que se han establecido precios de referencia. Si se determina que el nuevo medicamento no ofrece un beneficio incremental, pero tampoco cae en una clase terapéutica de precio de referencia, su precio está sujeto a negociación con la condición de que el precio negociado no exceda el de su medicamento de comparación.
En segundo lugar, el GBA elige las métricas que evaluarán el beneficio del nuevo medicamento. Estas métricas pueden diferir de las utilizadas por la EMA, el equivalente europeo de la Administración de Alimentos y Medicamentos de los Estados Unidos (FDA), en su revisión del medicamento para la autorización inicial de comercialización y para la cual el fabricante ha realizado ensayos clínicos.
En algunos casos, GBA ha rechazado métricas aceptables para la EMA, como la «supervivencia libre de progresión» para los medicamentos contra el cáncer, ya que los considera no relevantes para la calidad de vida del paciente. La supervivencia libre de progresión indica cuántos meses sobrevive el paciente después del tratamiento sin un aumento en el tamaño de sus tumores.
Esta métrica se correlaciona con la métrica de supervivencia general más importante, que indica el número de meses que el paciente permanece vivo después del tratamiento, pero a menudo no se correlaciona con la calidad de vida del paciente. En otros casos, GBA ha requerido que las empresas farmacéuticas proporcionan métricas que la EMA no requiere, principalmente indicadores de calidad de vida como el cambio en el dolor y las náuseas.
El GBA delega la evaluación clínica del nuevo fármaco en IQWiG,3 que considera la cartera de pruebas utilizadas para la autorización de comercialización por la EMA más otros estudios realizados por el fabricante. La evaluación final del beneficio del medicamento es decidida por el GBA. El GBA puede considerar que los fármacos ofrecen un beneficio mayor, sustancial, menor, positivo pero no cuantificable, o ningún beneficio incremental, en relación con el tratamiento de comparación. El beneficio no cuantificable se utiliza cuando se considera probable que el fármaco ofrezca un beneficio incremental, pero carece de pruebas suficientes para un juicio seguro de la escala.
Los medicamentos huérfanos, que a menudo no tienen un comparador directo y para los cuales la evidencia clínica puede basarse en muestras de pacientes muy pequeñas, generalmente reciben un beneficio no cuantificable. El GBA también evalúa la solidez de la evidencia disponible (débil, moderada o fuerte). El beneficio clínico de un fármaco puede ser reevaluado por GBA en respuesta a cambios en la evidencia disponible, lo que a veces desencadena una renegociación del precio.
Precios de referencia para productos que no ofrecen beneficios incrementales
Si el GBA considera que un medicamento no ofrece un beneficio incremental sobre los tratamientos existentes, generalmente lo asigna a una de las clases terapéuticas cubiertas por los precios de referencia. A los fabricantes se les permite establecer el precio que consideren apropiado para los medicamentos que caen en estas clases, pero la organización paraguas de aseguradoras de salud (GKV-SV) establece un límite a lo que las aseguradoras individuales contribuirán al pago. El GKV-SV establece su límite de pago cerca del percentil 30 en la distribución de precios dentro de cada clase terapéutica, lo suficientemente alto como para garantizar que los pacientes tengan más de una opción, pero lo suficientemente bajo como para garantizar que el pagador no sea responsable de pagar los precios más altos dentro de la clase. La mayoría de los medicamentos genéricos entran en el sistema de precios de referencia.
Aproximadamente el 34 por ciento de los medicamentos, el 80 por ciento de las recetas y el 33 por ciento del gasto en medicamentos en Alemania es para medicamentos sujetos a precios de referencia.4
Los pacientes deben pagar de su bolsillo la diferencia entre el precio establecido por el fabricante y el límite de reembolso basado en referencias establecido por la organización compradora. Muchos pacientes no están dispuestos a contribuir de su bolsillo y prefieren medicamentos con un precio por debajo del límite de referencia y sus médicos recetarán medicamentos en o por debajo del límite. De los productos sujetos a precios de referencia, aproximadamente el 84 por ciento tienen un precio de sus fabricantes igual o inferior al límite de precio de referencia y, por lo tanto, no están sujetos a costos adicionales compartidos.5 Estos productos representan el 92 por ciento de todas las recetas hechas para medicamentos de precio de referencia. Los fabricantes pueden presentar nuevos precios hasta dos veces al mes para los medicamentos en el sistema de precios de referencia. La organización paraguas de las compañías de seguros debe actualizar las clases terapéuticas cada trimestre y los límites de pago al menos una vez al año. Se permite a los fabricantes reducir sus precios hasta el límite de referencia para evitar la reducción inevitable del volumen de ventas; muchos lo hacen.
Para los medicamentos incluidos en el sistema de precios de referencia, los pacientes pueden estar obligados a pagar copagos adicionales, dependiendo del medicamento que seleccionen en consulta con sus médicos. Los pacientes que seleccionan un medicamento con un precio superior al máximo de referencia para su clase contribuyen con un copago más la diferencia entre el precio de su medicamento y el máximo de referencia. Estos copagos adicionales no cuentan para el máximo anual de costos compartidos de bolsillo de los pacientes. Sin embargo, los copagos adicionales son modestos, ya que la mayoría de los medicamentos incluidos en el sistema de precios de referencia son medicamentos genéricos más antiguos con precios típicamente bajos. Para los medicamentos no incluidos en el sistema de precios de referencia, las aseguradoras de salud alemanas requieren que los pacientes paguen solo el monto de los costos compartidos.
Aparte del requisito de que los pacientes paguen la diferencia entre el límite de referencia y el precio total de un producto, que solo se aplica en contextos en los que el paciente puede elegir una opción de bajo precio, Alemania impone límites estrictos a las responsabilidades financieras de bolsillo de los pacientes. El copago legal oscila entre un mínimo de 5 euros y un máximo de 10 euros por receta, hasta un máximo anual de bolsillo (para todos los servicios de atención médica) del 1 por ciento de los ingresos brutos para las personas con enfermedades crónicas y del 2 por ciento para otros. Aproximadamente una cuarta parte de los afiliados también tienen un seguro privado complementario, que cubre estos requisitos de costos compartidos.6
Precios negociados para productos que ofrecen beneficios incrementales
Si el GBA considera que un nuevo medicamento ofrece un beneficio incremental sobre los tratamientos existentes, se remite al GKV-SV para negociaciones de precios con el fabricante. La asociación paraguas de aseguradoras utiliza la evaluación del beneficio clínico de la GBA, así como los precios del medicamento de comparación, medicamentos terapéuticamente similares y precios cobrados en otras naciones europeas para negociar un descuento en el precio de lanzamiento del nuevo medicamento.
El GBA considera que algunos medicamentos no ofrecen un beneficio incremental, pero no entran en una clase terapéutica existente de precio de referencia, ya que debe haber al menos tres medicamentos terapéuticamente equivalentes para constituir una clase de precios de referencia. Estos medicamentos también tienen sus precios negociados entre el fabricante y la asociación de aseguradores, pero con la condición de que el precio del nuevo medicamento no pueda exceder el del producto de comparación elegido por el GBA.
Si las negociaciones entre la asociación paraguas de la aseguradora y el fabricante del medicamento no concluyen con un precio aceptable para ambas partes, el medicamento se remite a arbitraje. En este proceso, un panel de tres personas seleccionado por el fabricante, la organización de seguros y la GBA evalúa la evidencia y toma una decisión. Hasta finales de 2017, uno de los cinco (35 de 186) nuevos medicamentos evaluados por la GBA recibió un precio final a través del arbitraje en lugar de la negociación; para otros 24, las partes negociadoras llegaron a un acuerdo después de que se hubiera iniciado un proceso de arbitraje.7
Si un fabricante no puede obtener un precio aceptable ya sea a través de la negociación o el arbitraje, puede retirar su producto del mercado. Entre 2011 y 2017, 148 medicamentos fueron sometidos a una evaluación comparativa de la efectividad y sus precios fueron negociados por las aseguradoras y los fabricantes. De estos, 29 fueron retirados por el fabricante del mercado alemán en 2018.8 Para 12 de estos, el fabricante optó por retirar el producto inmediatamente después de los resultados de la evaluación de GBA, lo que se conoce como «exclusión voluntaria» del proceso de fijación de precios. En 16 casos, los medicamentos se retiraron en reacción al precio determinado, principalmente a través de arbitraje, y uno fue retirado porque su fabricante quebró.9
Lecciones para los Estados Unidos
El sistema alemán utiliza niveles modestos de costos compartidos como instrumento para influir en las elecciones de los consumidores de medicamentos con alternativas terapéuticamente equivalentes. Sin embargo, no aplica costos compartidos a los nuevos medicamentos que carecen de alternativas. Los precios de efectividad comparativa se utilizan para los nuevos medicamentos especializados que ofrecen beneficios clínicos sobre los tratamientos existentes.
Los fabricantes son libres de establecer los precios de sus productos, pero las aseguradoras no pagarán más por un nuevo medicamento que por sus competidores a menos que ofrezca un beneficio clínico adicional.
Para los medicamentos cubiertos por precios de referencia, el máximo de pago de las aseguradoras se establece en un nivel que garantiza suficientes opciones de opciones de bajo precio. Estos modelos ofrecen una alternativa al sistema estadounidense de fórmulas escalonadas.
En los Estados Unidos, el nivel de costos compartidos y la carga financiera resultante para los pacientes es alto, especialmente para los pacientes con afecciones médicas complejas.
Los pagadores estadounidenses a menudo imponen copagos modestos a los medicamentos de bajo costo con muchos sustitutos directos, pero un coseguro oneroso en medicamentos de alto costo con pocos sustitutos.
El coseguro no apunta al paciente hacia las opciones de medicamentos más rentables. Por el contrario, los diseños de seguros basados en precios de referencia identifican medicamentos que tienen un precio inferior al máximo de pago de la aseguradora y requieren solo un mínimo de costos compartidos.
2. This issue brief is based on several dozen interviews conducted in May and December 2017 with executives, analysts, and policymakers in Sickness Funds, pharmaceutical associations, the Ministry of Health, GBA, GKV-SV, universities, consulting firms, and nonprofit organizations active in the German health care system.
3. Treatments for orphan conditions are assessed in-house by GBA and are not delegated to IQWiG.
9. Product withdrawals are often driven by manufacturer fears that accepting a low price in Germany will lead to low prices in other European nations, 16 of which link their prices to those reported in the German market. If a product is withdrawn from the German market, its official price remains the one established unilaterally by the manufacturer at the time of launch. This list price is then used by other nations in international reference pricing comparisons, even if the drug in question is not in fact sold in Germany.
¿Cómo calcular el precio de referencia de los medicamentos? Fallo del Tribunal supremo en España.
La conclusión del Tribunal Supremo es que para calcular el precio de referencia de cada presentación debe estarse a la DDD de esa presentación que deberá tener en cuenta la potencia y eficacia de ésta pues esa dosis depende de la distinta farmacocinética y biodisponibilidad del medicamento. En otras palabras, el Alto Tribunal entiende que la DDD de la OMS puede no resultar aplicable en casos de medicamentos no equipotentes y que, entonces, deberá determinarse una DDD que tenga en cuenta las especificas características de tales presentaciones.
El resultado de la interpretación seguida por el Ministerio de Sanidad era que los medicamentos innovadores que con su formulación conseguían los mismos resultados que el resto de fármacos del conjunto con menos principio activo fueran más baratos. Ello suponía que los laboratorios cobraran menos por sus tratamientos que los medicamentos tradicionales
Dr. Carlos Alberto Díaz. Profesor Titular Universidad ISALUD.
Seguimos el hilo de publicaciones para ver lo que está haciendo un país que lo está haciendo muy bien, Israel, con disciplina, con el empleo de una estructura de seguridad ciudadana, usando sus sistemas de información. En argentina faltan testeos, para lograr limitar la transmisión, habilitar el autotest, incrementar la posibilidad de hacer el triple de las PCR, y el ritmo de la vacunación es muy lento, hay que hacer campañas muy fuerte y en todo el país y a todos sin otra limitación que lo que corresponda, a los adultos, seguir citaciones y convocatorias con los niños. No estoy en desacuerdo con un pase sanitario, en tanto, se genere conciencia de la importancia de la vacunación.
Los refuerzos aplastaron la ola delta de Israel. Ahora Está apostando a que una cuarta dosis hará lo mismo con ómicron.
La decisión de Israel esta semana de convertirse en el primer país en recomendar una cuarta dosis de vacuna para combatir la variante de ómicron altamente contagiosa se produjo después de que los funcionarios de salud concluyeron que un refuerzo inicial había cambiado el rumbo este otoño contra la variante delta.
Si bien reconocieron que su decisión no se basó en nuevos datos científicos sobre la variante ómicron, los funcionarios dijeron que pensaron que sería prudente recomendar una inyección adicional porque creen que la capacidad del refuerzo inicial para prevenir la infección ha ido disminuyendo con el tiempo.
La decisión, anunciada por funcionarios israelíes el martes, hará que una cuarta dosis, o segundo refuerzo, de la vacuna de Pfizer-BioNTech esté disponible para personas mayores de 60 años, personas con sistemas inmunológicos comprometidos y empleados en el sector de la salud. Todos los receptores elegibles tendrían que haber recibido su tercera dosis más de cuatro meses antes.
La decisión aún está a la espera de la confirmación del director del Ministerio de Salud, Nachman Ash, antes de convertirse en política nacional. Pero las instalaciones de todo el país se están preparando para comenzar a administrar la vacuna, y muchos dicen que están listas para comenzar el domingo.
El comité asesor nacional de coronavirus de Israel hizo su recomendación para la cuarta dosis el martes mientras aún recopilaba datos sobre la variante ómicron, diciendo que no tiene el lujo del tiempo. Se cree que la variante ómicron es tres veces más transmisible que las variantes anteriores, y aunque puede ser más leve (no ha causado aumentos masivos en las hospitalizaciones en el Reino Unido y las infecciones se han desplomado después de un aumento en Sudáfrica), muchos funcionarios de salud israelíes advierten que un aumento en los casos incluso moderados podría abrumar a los hospitales del país.
Según datos del Ministerio de Salud, 1.400 personas dieron positivo el miércoles por el coronavirus, el 45 por ciento de las cuales estaban completamente vacunadas. En Israel, eso significa que tenían al menos 12 años de edad y habían recibido una vacuna de refuerzo al menos una semana antes, o estaban dentro de los seis meses posteriores a haber recibido una segunda vacuna.
«Es un problema de gestión de riesgos», dijo Arnon Afek, subdirector del Centro Médico Sheba en el centro de Israel, quien es miembro de la sección de políticas del comité asesor nacional de coronavirus.
«El comité analizó la propagación increíblemente rápida de omicron, en el Reino Unido y otros países, y decidió que no tenemos tiempo. Decidieron que debíamos trabajar en paralelo: primero dar la recomendación de inmunizar, y luego ejecutar los estudios», dijo.
Cuando Israel lanzó una campaña durante el verano para las vacunas de refuerzo en respuesta a la variante delta, basó la decisión en estudios de Israel y del extranjero que muestran que la eficacia de la vacuna disminuyó significativamente en las personas en riesgo al menos cinco meses después de recibir la segunda dosis.
Los expertos coinciden en su mayoría en que los refuerzos, que se ofrecieron por primera vez a personas mayores de 60 años y grupos vulnerables en julio, luego a la mayoría de la población general en agosto, ayudaron a aplastar la ola de variantes del delta en Israel, permitiendo que la economía y las escuelas permanecieran abiertas. Atribuyeron a la vacuna, más que la aplicación del distanciamiento social, el cierre de las fronteras u otras medidas que solo ayudaron a retrasar la infección, el éxito de la estrategia.
En las semanas que siguieron, Clalit, el proveedor de salud más grande de Israel, descubrió que una tercera inyección de la vacuna Pfizer-BioNTech fue 92 por ciento efectiva para prevenir enfermedades graves y 93 por ciento efectiva para prevenir la hospitalización, en comparación con la disminución de la protección de solo dos dosis, según un estudio a gran escala publicado en la revista médica The Lancet en octubre.
Yasmin Maor, directora de la unidad de enfermedades infecciosas del Hospital Wolfson y miembro del comité asesor, dijo en una conferencia de prensa el miércoles por la noche que era «desafortunado» que el refuerzo pareciera proporcionar una protección insuficiente contra la variante omicron durante la «quinta ola» de Israel.
«Hubiéramos estado felices de tener un producto perfecto, pero tenemos que tener en cuenta cuántas vidas se salvaron porque administramos una tercera dosis de vacuna», dijo.
«Cuando el refuerzo era nuevo, había poca propagación de la infección. Cuanto más nos alejamos de ella, estamos viendo el doble de la tasa de infección», dijo Boaz Lev, quien encabeza el comité asesor, a los periodistas. «El precio será más alto si no vacunamos».
Israel ha confirmado al menos 341 casos de variantes de omicrones, la mayoría de los cuales fueron rastreados a viajeros que regresaron a Israel desde el extranjero.
El lunes, Israel agregó 10 países,incluidos Estados Unidos y Canadá, a su «lista roja» de destinos prohibidos, que también incluyen varios países europeos y casi toda África. La entrada de no ciudadanos está prohibida desde el mes pasado.
Amnon Lahad, presidente de medicina familiar de la Universidad Hebrea, dijo que la campaña de la cuarta dosis de Israel se basó en el «pánico», en lugar de la ciencia.
En lugar de seguir adelante con la próxima ronda de vacunas, dijo, Israel debería dedicar recursos a inocular a los aproximadamente 1 millón de personas no vacunadas del país, muchas de las cuales son miembros de las poblaciones desatendidas del país y carecen de fácil acceso a un centro de vacunación.
De una población de 9,3 millones, 6,4 millones han recibido su primera inyección, 5,8 millones la segunda y 4,1 millones su tercera, según el Ministerio de Salud.
Las ciudades con las tasas más bajas son las comunidades ultraortodoxas, árabes y de bajos ingresos.
Israel ha registrado más de 8.200 muertes relacionadas con el coronavirus. No ha confirmado ninguna muerte por la variante omicron.
El miércoles, el Ministerio de Salud dijo que un hombre de 65 años que murió el día anterior había contraído la variante delta, no la variante omicron como se informó originalmente.
Lahad criticó la política de Israel de clasificar la documentación de las reuniones del gabinete y otras discusiones gubernamentales sobre la política de coronavirus, diciendo que el «secreto» estaba contribuyendo a la vacilación de la vacuna en todo el país.
«Israel se está comportando como si estuviera en una guerra», dijo.
En primer lugar en el mundo, Israel se prepara para dar la 4ª vacuna COVID a más de 60 años, grupos de riesgo
Elogiando la recomendación del panel de expertos en salud, Bennett ordena el despliegue rápido de un refuerzo adicional de la vacuna para personas mayores de 60 años, inmunocomprometidos y trabajadores de la salud
Israel está listo para comenzar a ofrecer una cuarta vacuna contra el COVID-19 para mayores de 60 años, algunos grupos de riesgo y personal médico, en línea con una recomendación del martes de un panel de expertos en salud.
El primer ministro Naftali Bennett dio la bienvenida a la recomendación y ordenó que los funcionarios preparen una campaña para distribuir las vacunas, lo que significa que es probable que Israel se convierta en el primer país del mundo en implementar una cuarta dosis para ciertos grupos.
Informes no confirmados el miércoles dijeron que Israel comenzaría a ofrecer los disparos a partir del domingo 26 de diciembre.
Los expertos, un organismo gubernamental llamado Pandemic Treatment Staff, recomendaron que el personal médico, los adultos mayores de 60 años y otros grupos de riesgo reciban la cuarta dosis, después de esperar al menos cuatro meses desde la tercera dosis.
Entre el ocho y seis por ciento de los expertos del grupo favorecieron la medida.
La decisión necesita la aprobación final del Director General del Ministerio de Salud, Nachman Ash, antes de que entre en vigor.
«Esta es una noticia maravillosa que nos ayudará a superar la ola de Omicron que está envolviendo al mundo», dijo Bennett en un comunicado.
Bennett dijo que la inyección adicional también se administraría a los trabajadores de la salud.
Los ciudadanos de Israel fueron los primeros en el mundo en recibir la tercera dosis de la vacuna COVID-19, y seguimos siendo pioneros con la cuarta dosis también», dijo el primer ministro.
Israel fue pionero previamente en un tercer disparo de refuerzo, implementándolo para grupos en riesgo antes de expandir el programa a la población general semanas después, antes que gran parte del resto del mundo. Más de cuatro millones de israelíes han recibido la tercera dosis, de 9,3 millones de personas.
Sheba dijo que está llevando a cabo un estudio de la viabilidad de una 4ª inyección en un ensayo entre unos 200 voluntarios, examinando el efecto de los niveles de anticuerpos. El hospital dijo que era el primer ensayo de este tipo en el mundo y que se llevaría a cabo en cooperación con el Ministerio de Salud.
La profesora Galia Rahav, que dirige la unidad de enfermedades infecciosas de Sheba y forma parte del panel de expertos, dijo que la decisión de recomendar los refuerzos adicionales «no fue simple» dada la escasez de datos que muestran que la protección ofrecida por el tercer disparo estaba disminuyendo. «Pero al mismo tiempo. hay números terriblemente aterradores de lo que está sucediendo en el mundo en general», dijo a la Radio del Ejército, refiriéndose a la cepa Omicron de rápida propagación.
Los científicos aún no saben todo sobre Omicron, pero dicen que la vacunación debería ofrecer fuertes protecciones contra enfermedades graves y la muerte. Las cifras iniciales también han demostrado que la cepa es aún más contagiosa que la cepa Delta.
Bennett y el ministro de Salud, Nitzan Horowitz, han planteado la posibilidad de una cuarta dosis durante semanas,pero los expertos del Ministerio de Salud habían desaconsejado la medida, incluso en una decisión la semana pasada. Bennett dijo anteriormente que estaba esperando «impacientemente» que se aprobara la cuarta dosis, prediciendo que una nueva ola de infecciones era imparable.
La amenaza de Omicron ha estimulado nuevos llamados para que los israelíes se vacunen, incluidos los niños, por parte de funcionarios de salud y líderes gubernamentales.
El Ministerio de Salud dijo el martes que se habían confirmado 170 nuevos casos de la variante Omicron del coronavirus en Israel, duplicando el número de infecciones.
Ha habido 341 casos verificados de Omicron en Israel hasta la fecha, dijo el Ministerio de Salud en un comunicado. Otras 807 infecciones eran «altamente sospechosas» de ser casos de Omicron, pero estaban a la espera de verificación.
Los legisladores han renovado las restricciones para frenar las infecciones en los últimos días, incluidas las prohibiciones de viajes internacionales y la capacidad del lugar de trabajo público.
La vacunación contra el Covid 19 está evitando casos graves, disminuye las hospitalizaciones, pero no evita que transmita. Los casos en vacunados están resultando más leves. Los pacientes pueden aislarse en su domicilio. Se necesitarán que las nuevas vacunas logren Respuestas que aseguren más de un 80% de efectividad contra todas las variantes de preocupación en la humanidad, y sostener por más tiempo anticuerpos neutralizantes. Para ello desde las plataformas actuales deberemos ampliar los sitios de fabricación homologados y respaldados, para poder llegar a toda la humanidad con todas las dosis que se necesitan a menor precio. Se vuelcan tres artículos para conocer que dice el mundo científico. Empezamos con un artículo conceptual de Anthony Fauci sobre que es lo que define las característica de una vacuna universal. Si no se puede lograr una vacuna universal, otra que genere inmunidad duradera, nos tendremos que vacunar todos los años, y cuando aumenten los casos, es probable que sea necesarios refuerzos. Si esto ocurre así sería interesante utilizar la vacunación contra la influenza para co administrar contra el Covid 19.
Vacunas universales contra el coronavirus: una necesidad urgente
David M. Morens, MD, Jeffery K. Taubenberger, MD, Ph.D., y Anthony S. Fauci,
Los últimos 20 años la humanidad han sido testigos de cuatro brotes fatales de coronavirus: SARS (síndrome respiratorio agudo severo, 2002 y 2003), MERS (síndrome respiratorio de Oriente Medio, desde 2012) y ahora Covid-19 (desde 2019).
La evidencia científica y la realidad ecológica sugieren que los coronavirus volverán a surgir en el futuro, lo que podría representar una amenaza existencial. 1 Los betacoronavirus que causaron estas epidemias se distribuyen globalmente en numerosas especies de murciélagos. Se desconoce la extensión virológica y geográfica completa de este reservorio zoonótico; sin embargo, se ha extendido cada vez más a los seres humanos y otros mamíferos. 2
Debido a la conservación genética y estructural de los receptores entre las especies de mamíferos, muchos de estos betacoronavirus animales están “preadaptados” para infectar a los seres humanos al unirse a los receptores de la enzima convertidora de angiotensina 2 (ACE2), lo que facilita los desbordamientos virales y la transmisión continua. 3 Ya se han identificado algunos coronavirus animales que pueden tener potencial pandémico, y quedan muchos más por detectar.
Necesitamos un enfoque de investigación que pueda caracterizar el «universo coronaviral» global en múltiples especies, caracterizar la historia natural y la patogénesis de los coronavirus en animales de laboratorio y en humanos, y aplicar esta información en el desarrollo de vacunas «universales» ampliamente protectoras (que protegen contra todos los betacoronavirus , e idealmente todos los coronavirus).
En este punto, tenemos poca comprensión del universo de coronavirus endémicos y potencialmente emergentes. Aunque los coronavirus se distribuyen a nivel mundial, los puntos calientes de betacoronavirus más importantes se encuentran en el sureste de Asia y áreas contiguas del sur y suroeste de China. La identificación preliminar y la secuenciación de murciélagos y otros coronavirus adaptados a mamíferos de esta región revelan una evolución rápida y una enorme complejidad viral. Numerosas especies de murciélagos transmiten sarbecovirus (virus similares al SARS, incluido el SARS-CoV-2) entre sí y a numerosos mamíferos, incluidos los humanos, a un ritmo elevado. La generación de nuevos genomas a través de una infección mixta y una recombinación genética homóloga conduce a una diversidad genética coronaviral sustancial, análoga a la observada en la evolución del virus de la influenza A en aves silvestres, otros animales y humanos.
Para caracterizar completamente el ecosistema del coronavirus, un esfuerzo internacional de colaboración debe incluir un muestreo viral extenso de múltiples especies de murciélagos en múltiples lugares y de animales salvajes y de granja, incluidos los gatos de civeta de palma enmascarados ( Paguma larvata ) y los perros mapaches ( Nyctereutes procyonoides ), que con frecuencia son infectados con coronavirus, así como estudios virales y serológicos de humanos involucrados en el comercio de animales de granja y vida silvestre y aquellos que están expuestos ocupacionalmente a murciélagos. Dicho muestreo podría facilitar la identificación de una emergencia a tiempo para prevenir o controlar una pandemia; también permitiría el estudio de epítopos de reacción cruzada, que es importante para el desarrollo de vacunas, y respaldarían los estudios epidemiológicos y serológicos de la infección humana.
Para obtener información sobre la historia natural y la patogenia, será importante estudiar los coronavirus que probablemente alguna vez fueron pandémicos, pero ahora se han vuelto endémicos. Estos cuatro virus, los betacoronavirus OC43 y HKU1 y los alfacoronavirus 229E y NL63, causan principalmente infecciones respiratorias superiores leves y pueden estudiarse en animales de laboratorio y en humanos 4 para caracterizar su epidemiología, tropismo celular, respuestas inmunitarias provocadas, reacciones cruzadas y cruzadas. -epítopos protectores, y los mecanismos por los cuales sobreviven y evolucionan frente a una alta inmunidad poblacional. Los estudios de desafío ético en humanos 4 pueden realizarse utilizando herramientas genómicas, transcriptómicas e inmunológicas modernas.
Finalmente, necesitamos con urgencia vacunas universales contra el coronavirus. 5 En los Estados Unidos, la pandemia de Covid-19 ha sido parcialmente controlada por medidas estándar de salud pública como el distanciamiento social, el enmascaramiento, el aislamiento de personas enfermas y expuestas, el cierre de lugares donde las personas se congregan en lugares cerrados y otras medidas, así como por Vacunas SARS-CoV-2 (dos vacunas de ARN mensajero y una vacuna vectorizada por adenovirus). Sin embargo, por importantes que sean estas vacunas, su eficacia protectora disminuye con el tiempo, lo que requiere dosis de refuerzo. La vacunación tampoco ha sido capaz de prevenir infecciones «irruptivas», lo que permite la transmisión posterior a otras personas incluso cuando la vacuna previene enfermedades graves y mortales.
Las personas que han sido infectadas naturalmente con SARS-CoV-2 también pueden re-infectarse, como se ha demostrado con coronavirus endémicos, virus de influenza, virus respiratorio sincitial (RSV) y muchos otros virus respiratorios. Además, la inmunidad que sigue a la infección natural por el SARS-CoV-2, combinada con la inmunidad inducida por la vacuna, hasta ahora no ha impedido la aparición y rápida propagación de variantes virales como la variante delta altamente transmisible (B.1.617.2) y la reciente identificó omicron (B.1.1.529) «variante de preocupación», que a fines de noviembre, parecía ser altamente transmisible. Se desconoce si se puede lograr una inmunidad protectora permanente y cómo, y si puede prevenir la aparición de variantes de escape inmunológico del SARS-CoV-2.
Estos hechos aleccionadores sugieren que es poco probable que el SARS-CoV-2 sea eliminado, y mucho menos erradicado; probablemente seguirá circulando indefinidamente en brotes periódicos y endémicos. Mientras tanto, un número desconocido de coronavirus animales, de transmisibilidad y letalidad desconocidas, pueden surgir en el futuro previsible. Por lo tanto, debemos acelerar enormemente nuestros esfuerzos en la vacunación contra el coronavirus.
Las limitaciones de las vacunas contra el SARS-CoV-2 sugieren que, en última instancia, deberán ser reemplazadas por vacunas de segunda generación que induzcan una inmunidad protectora más amplia y más duradera. Ahora debemos priorizar el desarrollo de vacunas de protección amplia como las vacunas universales contra la influenza en las que hemos estado trabajando en los últimos años. Una vacuna universal contra el coronavirus protegería idealmente contra el SARS-CoV-2 y los muchos coronavirus de origen animal que podrían causar futuros brotes zoonóticos y pandemias. Las características ideales de tales vacunas incluyen propiedades asociadas con la protección tanto individual como comunitaria en caso de pandemia.
PROPIEDADES IDEALES DE UNA VACUNA UNIVERSAL CONTRA EL CORONAVIRUS.
PROTECCIÓN INDIVIDUAL
Necesario
Previene la enfermedad clínica
Previene la infección por todos los sarbecovirus y merbecovirus.
Previene la infección por derivación viral y variantes de recombinación.
Provoca una respuesta inmune rápida y robusta
No tiene inmunogenicidad limitada de la vacuna en personas con inmunidad preexistente
Induce inmunidad a múltiples componentes virales.
Es seguro y aceptable para el público.
Es seguro para mujeres embarazadas.
No induce una mejora dependiente de anticuerpos con la posterior exposición al virus de tipo salvaje
Se puede utilizar en personas de todas las edades.
Deseable
Es muy eficaz en una dosis.
Induce una sólida inmunidad sistémica de por vida
Induce una sólida inmunidad de la mucosa de por vida
Induce un aumento de la inmunidad con la posterior exposición al virus de tipo salvaje
No altera el microbioma respiratorio
Es asequible y se puede utilizar en países de bajos ingresos.
Es eficaz en personas con inmunosupresión.
PROTECCIÓN COMUNITARIA
Necesario
Cubre todos los sarbecovirus y merbecovirus
Cubre todos los coronavirus humanos endémicos
Puede usarse para la prevención de una pandemia
Se basa en una plataforma que se actualiza fácilmente con nuevos antígenos.
Deseable
Evita la transmisión
Reduce o acorta la diseminación viral.
Crea inmunidad de grupo duradera
No provoca la neutralización de mutantes de escape.
Es estable en almacenamiento
Induce un impulso en la protección inmunológica con vacunación secuencial
* Las características enumeradas describen una vacuna verdaderamente universal, aunque es poco probable que los enfoques de vacunas actuales logren todos estos objetivos. La máxima prioridad debe ser la cobertura universal de betacoronavirus, con cobertura adicional de coronavirus endémicos y otros.
Otro Artículo.
En las últimas dos décadas, hemos tenido tres brotes importantes de enfermedades infecciosas humanas causadas por coronavirus zoonóticos: SARS en 2002-2003, síndrome respiratorio de Oriente Medio (MERS) desde 2012 y Covid-19 desde diciembre de 2019. 14 Los tres brotes causaron devastadores pérdidas humanas y económicas en todo el mundo. Para el coronavirus SARS-CoV-1 y MERS, todavía no tenemos una vacuna autorizada. Para el SARS-CoV-2, la velocidad sin precedentes del desarrollo de la vacuna ha dado como resultado varias vacunas autorizadas para la vacunación masiva en humanos. 15
La reciente aparición de variantes del SARS-CoV-2 que pueden evadir parcialmente la respuesta inmune a las vacunas que se basan en la cepa del virus original 16-18 aumenta la necesidad de una vacuna contra el coronavirus de segunda generación que proteja contra la infección por todos los conocidos y cualquier variante futura del SARS-CoV-2. Se están realizando esfuerzos para desarrollar una vacuna de este tipo. 19
Sin embargo, una vacuna “de ensueño” cubriría no solo el SARS-CoV-2 y sus variantes conocidas de preocupación, sinotambién las variantes futuras de preocupación y otros coronavirus con potencial conocido de causar enfermedades humanas graves en el futuro, 20 muy probablemente del género betacoronavirus. . 3 Aunque se han hecho llamamientos para desarrollar vacunas contra el pan-betacoronavirus o el pan-coronavirus, 20 un objetivo más realista y urgente es una vacuna contra el pan-sarbecovirus o una vacuna contra el coronavirus de tercera generación. Primero, dada su alta transmisibilidad en humanos, los sarbecovirus presentan un riesgo más alto que los otros coronavirus zoonóticos no sarbecovirus, a pesar de que el MERS tiene la tasa de letalidad más alta entre las enfermedades causadas por estos virus. 14En segundo lugar, todos los sarbecovirus zoonóticos identificados hasta la fecha utilizan hACE2 como receptor de entrada, lo que garantiza una mayor probabilidad de éxito en la generación de una vacuna contra el pan-sarbecovirus al inducir anticuerpos de neutralización cruzada que bloquean la interacción común entre hACE2 y virus. Un estudio de mutagénesis profunda de varios RBD de sarbecovirus indicó limitaciones mutacionales en el plegamiento y la unión de ACE2. 4 Estas limitaciones pueden explicar la existencia de epítopos neutralizadores de clado cruzado altamente conservados, pero no inmunodominantes, en los RBD de sarbecovirus. Este hallazgo también apunta a un posible candidato a vacuna de pan-sarbecovirus que inducirá fuertes anticuerpos neutralizantes de cruzado dirigidos a los epítopos más conservados que desempeñan un papel en la neutralización del virus, ya sea que estén ubicados dentro o fuera de la interfaz directa RBD-ACE2.4
EFECTO DE CROSS-CLADE PRIME Y BOOST
Planteamos la hipótesis de que los anticuerpos neutralizantes del pan-sarbecovirus observados en las muestras de suero vacunadas contra el SARS-CoV-1 eran el resultado del enriquecimiento de anticuerpos neutralizantes de reacción cruzada de una cebado cruzado (infección) y un refuerzo (vacunación). Para probar esta hipótesis, utilizamos dos enfoques diferentes. En primer lugar, realizamos un estudio de competición utilizando el anticuerpo monoclonal 5B7D7, que se sabe que se une a los 10 RBD y es capaz de neutralizar los 10 sarbecovirus (Fig. S2A). Los datos que se muestran en la Fig. S2B indican claramente que el panel vacunado contra el SARS-CoV-1 es el único grupo de muestras de suero que mantuvo un alto nivel de inhibición contra todos los virus, lo que confirma los hallazgos que se muestran en las Figuras 1A y 1B. Además, la potenciación de los anticuerpos neutralizantes de reacción cruzada en las muestras de suero vacunadas contra el SARS-CoV-1 se confirmó aún más mediante la tinción directa de las células B con el uso de proteínas RBD específicas del virus. Como se muestra en la Figura 1C y 1D , las células B teñidas doble (es decir, las células B que se unen a los RBD tanto del SARS-CoV-1 como del SARS-CoV-2) se enriquecieron significativamente en el panel vacunado contra el SARS-CoV-1 como en comparación con los paneles de pacientes sanos y vacunados contra el SARS-CoV-2.
Los hallazgos del estudio actual mostraron la inducción eficiente de anticuerpos neutralizantes de pan-sarbecovirus de alto nivel y de amplio espectro que pueden neutralizar todas las variantes de interés y cinco sarbecovirus preemergentes. Nuestros hallazgos mostraron la viabilidad de lograr la neutralización del pan-sarbecovirus a través del refuerzo de clados cruzados. Estudios anteriores han proporcionado datos de prueba de concepto para una vacuna contra el pan-sarbecovirus en estudios en animales. 21-23
En comparación, nuestro estudio mostró un aumento de los anticuerpos neutralizantes del pan-sarbecovirus en humanos que fue más uniforme y más fuerte que el observado en animales. Un nivel tan alto de refuerzo de anticuerpos neutralizantes de pan-sarbecovirus puede lograrse con una vacuna de refuerzo cruzada de dosis única, como se indica con el suero de dos de los ocho participantes de nuestro estudio.
El sVNT multiplex recientemente desarrollado que se utiliza en este estudio puede desempeñar un papel fundamental en los estudios que requieren una comparación exacta de los niveles de anticuerpos neutralizantes contra diferentes virus. Esto es especialmente importante cuando no todos los virus vivos están disponibles, como es el caso de murciélago coronavirus RaTG13, 1 bate de coronavirus WIV1 y RsSHC014, 13 y pangolines coronavirus GD-1 y GX-P5L. 12 Aunque somos conscientes de que existen anticuerpos neutralizantes que se dirigen a regiones no RBD, 24 múltiples estudios han demostrado que para los sarbecovirus, la mayoría de los anticuerpos neutralizantes se dirigen al RBD inmunodominante. 4,25Estudios anteriores han indicado que la sVNT basada en RBD tiene una alta concordancia con las pruebas de neutralización basadas en virus vivos. 5,8 Hemos proporcionado más datos sobre la concordancia entre el sVNT y una prueba de neutralización de virus pseudotipados para tres sarbecovirus seleccionados.
Los datos actuales en humanos se obtuvieron de supervivientes de la infección por SARS-CoV-1 inmunizados con una vacuna de ARNm basada en SARS-CoV-2 S. Se necesita más investigación para determinar si la administración en serie de vacunas basadas en dos proteínas S o RBD relacionadas lejanamente en el orden inverso, es decir, cebado del clado SARS-CoV-2 seguido de refuerzo del clado SARS-CoV-1 producir un nivel similar de anticuerpos neutralizantes de pan-sarbecovirus. Si tiene éxito, esto sentará una base sólida para el desarrollo de una vacuna Covid-19 de tercera generación para controlar las variantes actuales y emergentes de preocupación, así como para prevenir futuras pandemias de sarbecovirus.
El futuro de la vacunación contra el SARS-CoV-2: lecciones de la influenza
Arnold S. Monto, MD
Después de un período de caída de las tasas de enfermedad por Covid-19, la reciente propagación de la variante delta del SARS-CoV-2 fue una gran decepción y requirió un nuevo examen de algunas suposiciones anteriores. Esta reconsideración puede ser, al menos en parte, una corrección a las opiniones demasiado optimistas de lo que podrían lograr las vacunas contra el SARS-CoV-2 altamente efectivas. Algunos observadores esperaban que las vacunas pudieran eliminar la transmisión del virus, el objetivo final de alcanzar la inmunidad colectiva. 1 Una imagen más probable de nuestro futuro con este virus se enfoca si examinamos los patrones de infección bien conocidos de otro virus respiratorio, la influenza, tanto dentro como fuera de las pandemias. Esa experiencia puede ayudarnos a restablecer las expectativas y modificar los objetivos para tratar el SARS-CoV-2 a medida que se adapta aún más en la propagación global.
Los primeros resultados de los ensayos clínicos y los estudios observacionales de las vacunas de ARNm contra el SARS-CoV-2 indicaron que no solo eran muy eficaces para prevenir la infección sintomática, sino que también lo eran para prevenir la infección asintomática y, por lo tanto, la transmisión. 2 El criterio básico utilizado para la autorización de uso de emergencia por la Administración de Alimentos y Medicamentos fue uno estándar: la prevención de la infección clínica confirmada por laboratorio que cumple con una definición de caso. El efecto sobre las infecciones asintomáticas fue una grata sorpresa, porque se pensó que la mayoría de las vacunas para enfermedades respiratorias, incluida la influenza, tienen «fugas», es decir, permiten cierto grado de infección asintomática y son mejores para prevenir la infección sintomática.
Los datos iniciales sobre la infección inaparente por SARS-CoV-2 fortalecieron la esperanza de que, a un cierto nivel de vacunación, la transmisión cesara por completo. Para muchos de nosotros, esta esperanza parecía demasiado optimista, y lo parece aún más ahora; la variante delta altamente transmisible causa infecciones asintomáticas y, a veces, enfermedades (aunque generalmente leves) en las personas vacunadas, probablemente debido a un mayor potencial de crecimiento, así como a la disminución de la inmunidad, que también implica la disminución de los niveles de anticuerpos IgA.
La eliminación de una enfermedad por medio de la inmunidad colectiva funciona mejor cuando el agente tiene baja transmisibilidad y requiere la ausencia de focos de personas susceptibles. La eliminación de Covid-19 parecía teóricamente posible, porque el virus del SARS original de 2002 finalmente desapareció. Ese virus, sin embargo, no transmitió tan bien como la cepa inicial de SARS-CoV-2. Ocurrió en regiones limitadas y se caracterizó por una propagación focal, incluidos eventos de super propagación. Este patrón, que también se observó en los primeros días del SARS-CoV-2, se denomina “sobredispersión”: el 10% de los casos, por ejemplo, puede ser responsable del 80% de la transmisión.3 Estas dinámicas explican por qué hubo grandes diferencias en la prevalencia de anticuerpos dentro de una ciudad determinada y una dispersión global irregular al principio de la pandemia. Se pensaba que la sobredispersión era un rasgo inestable que desaparecería, y que la transmisión se volvería más uniforme y más alta en general. Esa transición parece haber ocurrido cuando las nuevas variantes se hacen cargo.
Dado el desfile de variantes, su variabilidad de transmisibilidad y la preocupación constante por los cambios antigénicos que afectan la protección de la vacuna, creo que ahora debería quedar claro que no es posible eliminar este virus de la población y que deberíamos desarrollar planes a largo plazo para tratar con él después de que las sobretensiones insoportables estén completamente controladas. La influenza pandémica y estacional proporciona los modelos más apropiados para ayudar en el desarrollo de estrategias en el futuro.
Al igual que con el SARS-CoV-2, cuando aparece una nueva cepa de influenza pandémica, su propagación puede abrumar al sistema de atención médica. Las olas de infección atraviesan una ciudad en semanas y un país en meses, pero hay poca evidencia de que ocurran eventos de superpropagación. A partir de entonces, el virus pandémico persiste como una nueva cepa estacional y se producen cambios antigénicos, aunque probablemente no tan rápido como vemos con el SARS-CoV-2. La nueva cepa se une a los otros tipos y subtipos de influenza estacional que reaparecen cada año. El objetivo de la vacunación es controlar los brotes inevitables y reducir las tasas de enfermedad y muerte de moderadas a graves.
La prevención de enfermedades leves, aunque importante, es menos crítica.
Resumen del proceso de selección de virus de la Organización Mundial de la Salud (OMS) para las vacunas anuales contra la influenza.
La readministración de la vacuna contra la influenza se ha convertido en un evento anual para gran parte de la población, en respuesta tanto a la disminución de la inmunidad como a la aparición de variantes, denominadas deriva antigénica, que requieren vacunas actualizadas. Incluso cuando no hay una deriva sustancial, se recomienda la revacunación debido a la disminución de la inmunidad. Pero la deriva antigénica es un problema constante y se monitorea a nivel mundial, con la composición de la vacuna actualizada a nivel mundial dos veces al año sobre la base de las recomendaciones de una consulta de la Organización Mundial de la Salud. 4 Como se indica en la tabla, se tienen en cuenta varios criterios en las decisiones sobre qué cepas incluir en las vacunas. La eficacia de la vacuna contra la infección sintomática confirmada por laboratorio nunca supera el 50 al 60% y, en algunos años, es mucho menor. Por lo tanto, el valor de las vacunas contra la influenza, que ahora se administran al 70% de las personas en algunos grupos de edad, no radica en eliminar los brotes, sino en reducirlos y prevenir complicaciones graves.
Aunque puede haber similitudes entre el SARS-CoV-2 y la influenza, también existen diferencias significativas. La diferencia más obvia es la eficacia de las vacunas contra el SARS-CoV-2, que actualmente es mucho mayor de lo que podemos lograr con las vacunas contra la influenza. Si ese grado de eficacia continuará es una de las muchas preguntas abiertas que solo pueden responderse con el tiempo. Sin embargo, está claro que la revacunación será necesaria, por las mismas razones por las que es necesaria la revacunación contra la influenza: variación antigénica y disminución de la inmunidad. Los datos sobre la frecuencia de reinfección con coronavirus estacionales pueden no ser relevantes, pero sugieren que la protección es relativamente a corto plazo incluso después de una infección natural. 5 Será necesario determinar la frecuencia y las consecuencias de la revacunación.
Esperemos que ciertos problemas con la vacuna antigripal -como el fracaso de la vacunación, en algunos años, para producir el aumento deseado de protección en personas previamente vacunadas- no ocurran con las vacunas SARS-CoV-2. Deberán abordarse otras cuestiones, como la variante a la que se dirigirán las vacunas. La exitosa colaboración público-privada en la selección de cepas de influenza ofrece un modelo para abordar estos problemas. Las vacunas contra el SARS-CoV-2 se utilizarán a nivel mundial, y la cepa o cepas contenidas en las futuras vacunas deberán elegirse a nivel mundial, en consulta con los fabricantes.
La mayoría de las predicciones sobre la forma del mundo posterior a Covid-19 han sido inexactas, un reflejo de los rápidos cambios en el conocimiento. Pero ahora podemos ver una imagen emergente en la que el uso de vacunas eficaces seguirá siendo fundamental a largo plazo. No obstante, seguirá siendo posible un aumento de las infecciones asintomáticas y las enfermedades leves en las personas vacunadas, a medida que sigan surgiendo variantes. El recuento de hospitalizaciones y muertes puede ser más importante para monitorear el impacto general que el número de casos, siempre que las vacunas sigan siendo en gran medida eficaces para prevenir enfermedades graves. La posibilidad de enfermedades graves en una pequeña proporción de personas vacunadas enfatiza una de las mayores necesidades insatisfechas que enfrentamos actualmente: énfasis continuo en mejores agentes terapéuticos y antivirales,
El calendario y la composición futuros de las dosis de vacuna de refuerzo deberán determinarse sobre la base de estudios observacionales. Actualmente disponemos de pocos datos sobre las vacunas sin ARNm, en particular las vacunas a base de proteínas, que pueden tener características diferentes a las de las vacunas de ARNm, especialmente en términos de duración de la inmunidad.
En general, la situación será inestable, pero necesitaremos el uso continuo de vacunas para evitar consecuencias graves, incluso si las enfermedades más leves siguen ocurriendo con poca frecuencia. Necesitamos aprender a vivir con estas enfermedades, al igual que hemos aprendido a vivir con la influenza.
Esta era del conocimiento y cristalización de la inequidad:
Estamos viviendo una nueva era, que en sentido crítico no es una nueva normalidad. Estrictamente lo anterior a la irrupción de la pandemia, no era una normalidad. Se constituía como una Desigualdad injusta creciente, concentración de la riqueza inadecuada, alteración de los valores de la sociedad, deterioro de la gobernanza, perdida de las libertades por populismos y exclusión, vidas invadidas por algoritmos de consumos y preferencias, datos e información, constituyeron una erupción volcánica desordenada de mala calidad, no conocimiento, ni tiempo para competencia, que no nos dejaba pensar, que nos hacía actuar por las corrientes de los intereses, no del conocimiento, ni de la verdad, si alguna vez coinciden verdad y conocimiento bueno, puede ser, pero primero son las ventas, es la facturación, la velocidad de retorno de capital invertido, las utilidades, aparecen monedas sin respaldo, como las cripto, que son una entelequia sin valor, en oposición a un país que financia EE.UU. su déficit con papeles de color verde que quiere el resto del mundo, como reserva de valor y que como contrapartida, ofrece protección, con un poderío militar itinerante, inigualable, erigiéndose en el gendarme del mundo, que lleva a los aliados de la OTAN a justificar sus guerras en Afganistán, siria, irak, etc, para vender protección y hacernos creer que con ellas consolidan la «paz mundial», el objetivo se constituye en pos del predominio de occidente, suma almas como tropas, que juegan guerras que no les pertenecen, que luego lloran o vuelven discapacitados y olvidados a sus países, esta potencia dice con quién hay que aliarse, comerciar, intercambiar, es como el gran imperio del siglo XXI, en tanto, en nuestra realidad de los sistemas de salud, con un escenario de riesgo para la sustentabilidad, el complejo industrial médico, surgieron entidades supranacionales, empresas que son estados, que son los laboratorios farmacéuticos y las fábricas de dispositivos médicos. Empresas que crecen nutriéndose del conocimiento que surge de las Universidades públicas, que incuban negocios de las investigaciones que son grandes oportunidades de rentabilizar inversiones, más que con la industria armamentística.
Con la fortaleza del conocimiento del genoma humano y su secuenciación, terapias génicas y celulares, inmunoterapia celular, simulación, impresión 3 D, cirugía robótica, inteligencia artificial, sensores o dispositivos portátiles avanzados, de técnicas de hibridización, delección, identificación de anticuerpos monoclonales, con el desarrollo de nanopartículas, que encapsulan material genético, se convierten en proveedores de bienes de un mercado global, cuyo alcance depende de que se pueda pagar, y sino exigir, que se garantice con bienes soberanos, un fondo de garantía para evitar las demandas por lanzar vacunas al mercado con aprobación parcial.
Los ciudadanos del mundo no están sanos, sino pre-enfermos o insuficientemente diagnosticados.
Estas imperfecciones afectan el desarrollo de la era del conocimiento, porque el mismo es el combustible nuevo para los clientes habitantes del planeta, que no están sanos, sino pre enfermos de algo, o insuficientemente diagnosticados. los que no tienen recursos para pagar semejante lujo, como es tener servicios de salud equitativos, e integrados, es un bien suntuario, los pobres de américa latina, del África, de las ciudades, no tienen derecho a estar enfermos, o bien se enfermarán de la transición demográfica y epidemiológica, morirán de paludismo, de VIH, de hepatitis C, de Cáncer no diagnosticado, de hambre, de ébola, o de Covid 19, que no igualó nada. Derrochando años de vida y proyectos, que seguiremos denominando años de vida potencialmente perdidos. Los sistemas de salud no tienden naturalmente a la equidad. No solo quienes trabajamos en el, podemos intentar mitigar su tendencia a cristalizar la desigualdad, sino los gobernantes y los que tienen responsabilidades políticas. Sabiendo que es poco lo que podemos hacer, aunque lo seguiremos intentando, haciendo lo que podamos en nuestra pequeña aldea, nuestro pequeño lugar. Superando el enfoque reduccionista de no saber qué es salud, solo preocuparnos por la enfermedad, la cronicidad, la multimorbilidad, la probabilidad, y los factores de riesgo.
Empresas Farmacéuticas supranacionales contra el Nihilismo médico, generando halcones de la prescripción:
Están radicadas en alguna Nación, casualmente, pero no pertenecen a ella. No son embajadas, porque no tienen territorio. sino mercados. Su producción es la renta, si producen renta, ganancias, no la salud, han descubierto que deben impulsar a los halcones contra el nihilismo médico, para que prescriban en una actitud acrítica lo que la ciencia intencionada le dice que es lo mejor, aunque más costoso. Un intervencionista halcón tiene pocas dudas sobre la eficacia de un nuevo tratamiento y lo adopta rápidamente en la práctica.
Los productores innovan diariamente en la forma de influenciar en los prescriptores médicos para que se conviertan en halcones. Constituyendo sesgadamente la evidencia. Forzando en los Hawkish la prescripción intencionada. Creciendo año tras año el monto de la facturación y el valor de las empresas, avanzados sobre otras inversiones que requiere la sociedad y que le dan más salud. Hiper Valorizando la tecnología médica y específicamente los medicamentos, en detrimento de otras condiciones de vida, de los determinantes sociales de la salud, que permitirán generar libertad, oportunidades y ascenso social. Mejorar la desigualdad y los determinantes sociales no es negocio.
No se puede negar los avances de la tecnología farmacéutica, tanto la terapèutica diagnóstica y vacunal, lo que son maledicentes son la inequidades sobre las cuales encuentran su base de distribución, que la despoja de cualquier componente humanitario.
Estos avances, estas nuevas tecnologías disruptivas ¡han contribuido a la curación de muchas enfermedades es innegable, pero no justifica esta voracidad actual, avance que no tiene simetrías, constituyéndose como abuso de una posición dominante, por la posesión de un bien, que todos desean y no pueden adquirir, un bien, que por monopolio concedido, no pueden competir.
Qué no ceden ni siquiera ante la fuerza de la principal potencia mundial, y de ningún país soberano o tratado o acuerdo impulsado por la Organización Mundial de la salud.
Entonces se debe adoptar una política desafiante desde la soberanía nacional, de intentar instalar la mayor cantidad de tecnologías, que puedan sustituir estos comportamientos monopolísticos, que condicionan la distribución de bienes de salud entre quienes lo pueden pagar, no entre quienes lo necesitan.
La salud es un componente sustancial del capital humano y un factor determinante en la generación de crecimiento económico sostenible.
La buena salud reduce la cantidad de horas perdidas debido a enfermedad, incrementa la productividad de la fuerza de trabajo, aumenta las rentas individuales, por ello produce reasignación inmediata de la riqueza. Las epidemias y las endemias erosionan la cooperación social, la estabilidad política y económica. Como consecuencia de ello es dable sostener que la salud integra el sistema de derechos inalienables. El enfoque de derechos entendido como un sistema coherente de principios y pautas aplicables a las políticas de desarrollo, fija marcos para la definición de los contenidos y las orientaciones, pero también incide en la elaboración e implementación de las políticas. El sistema de salud, sus integrantes deben ser innovadores disruptivos, como el sector informático. Produce servicios calificados de mano de obra intensiva. Muy pocas fábricas tienen hoy más de mil empleados, muchos hospitales tienen cifras superiores. Este manejo fenomenal de recursos e inflacionario vegetativamente, crece el 7% por año, que implica que los estados tengan que invertir más que el crecimiento del producto bruto a Salud. Por ello los sistemas universales se limitan por lista de espera, y los organizados por planes de salud, como el nuestro, por su capacidad de pago.
En la actualidad los pacientes exigen a la Medicina, más de lo que esta puede darle y creen siempre que las cosas no marchan bien, se debe que el profesional que tiene enfrente es un médico que no está informado, o que no está accediendo a la solución de su problema por cuestiones económicas. Pero cuidado más dinero en la salud, no es igual a mejores servicios y más equitativos. La inversión en salud es un prerrequisito para el crecimiento económico, por lo tanto debe estar incentivada, y no como en la actualidad. Desde las naciones desarrolladas con sistemas universales de salud, financiados por impuestos estas distorsiones también se conocen y se viven, entonces los ciudadanos empiezan a pagar por elegir otro médico, por acceder antes al especialista que lo que determina su lista de espera, o en intervenciones de corta estancia a mayor confort hotelero. afectando la equidad del sistema, por lo cual también forjaron el desarrollo de esas naciones.
Conclusión:
En Argentina tener un servicio de salud que te permita formalizar tu cobertura, nominalizarla y georeferenciarla es una aspiración de sus habitantes, que si bien pueden acceder al hospital, la prelación está concedida por la gravedad, presionando las salas de emergencia, o esperando ser atendido desde filas organizadas desde la madrugada.
Los servicios de salud, se financian con aportes y contribuciones, que una parte de ellos es un salario diferido, que los trabajadores valorizan mucho, inclusive cuando acceden a un empleo.
Pero esta forma de financiamiento en un mercado competitivo de seguros esta en crisis, no alcanza con la recaudación, para cubrir el costo del programa y además está inadecuadamente distribuida, con inadecuados subsidios cruzados.
Carlos Alberto Díaz. Profesor Titular Universidad ISALUD. 2021. 24 de Diciembre.
La importancia de este trabajo que se detalla en esta intervención, fue Publicado recientemente en Lancet, por Toback S. et al, 2021, expresa que la administración conjunta de la vacuna contra la influenza con una dosis de cualquier programa de vacunas contra el COVID-19, o con una dosis de refuerzo posterior de la vacuna contra el COVID-19, podría superar cualquier posible interferencia inmunitaria.
Este subestudio es el primero en mostrar la seguridad, inmunogenicidad y eficacia de cualquier vacuna COVID-19 cuando se administra conjuntamente con una vacuna contra la influenza estacional o cualquier otra vacuna.
Esta es la reproducción de un trabajo de investigación alentador en fase 3, con una vacuna Novavax contra el Covid 19 y otra Vacuna contra la influenza o Gripe.
Es una alternativa, opción, y oportunidad importante, fundamental, para contener la transmisión de Covid e influenza y proteger a la población, este trabajo demuestra que es seguro, es efectivo. Servirá si se puede co-producir vacunas en diferentes partes del mundo y el precio es asequible.
Esto requiere otra visión, que el Mundo, que los países desarrollados no tienen, no se está trabajando para terminar con la pandemia, en forma de una estrategia global. Lo demuestra el fracaso del programa Covax, en el cual se había depositado una esperanza muy fuerte. Cada país hace lo que supone es conveniente para sí. No entiende que los países tienen fronteras abiertas, que el virus viaja a 800 km por hora, que a los pocos días de estar en una persona en Sudáfrica, está en Gran Bretaña, en New York, en cualquier parte del mundo. Los confinamientos sirven para permitir que los sistemas de salud se preparen, no colapsen, tenga camas y respiradores. Hace dos años que no circula el virus de la influenza, entonces, todos querrán vacunarse contra el covid, dándole una oportunidad a la diseminación de la influenza, y si el virus que circula fuere de otra cepa muy diferente podrá causar otra epidemia como el 2009. Lo digo, pensando en función de lo que leo para escribir estos materiales, quisiera que la comunidad científica lo debata, lo piense y se lo informe a los políticos, para no correr otra epidemia, muy de atrás. Con muchas muertes evitables en nuestra mente.
Intervención.
Seguridad, inmunogenicidad y eficacia de una vacuna contra la COVID-19 (NVX-CoV 2373) co-administrada con vacunas contra la influenza estacional: un sub-estudio exploratorio de un ensayo aleatorizado, ciego al observador, controlado con placebo, de fase 3.
Aún no se ha informado del perfil de seguridad e inmunogenicidad de las vacunas contra la COVID-19 cuando se administran concomitantemente con las vacunas contra la influenza estacional. Por lo tanto, nuestro objetivo fue informar los resultados de un subestudio dentro de un ensayo de fase 3 en el Reino Unido, mediante la evaluación de la seguridad, la inmunogenicidad y la eficacia de NVX-CoV 2373 cuando se administró conjuntamente con vacunas contra la influenza estacional autorizadas.
Desarrollo:
Los esfuerzos mundiales de vacunación contra la COVID-19 ya están en marcha con más de 6.900 millones de dosis de vacunas administradas hasta el 2 de noviembre de 2021.1
Este programa continuo de vacunación masiva contra la COVID-19 sin duda coincidirá con los programas de vacunación contra la gripe. Con el inicio de las campañas de refuerzo y la continuación de la vacunación en serie primaria, el momento de tales dosis probablemente se superpondría con la temporada de influenza 2021-22 en muchos entornos. Actualmente, no existen datos para la administración conjunta de vacunas covid-19 con otras vacunas, ya que la mayoría de los ensayos de fase 3 de vacunas COVID-19 excluyeron a los participantes con recepción reciente o planificada de otras vacunas autorizadas o requirieron un intervalo de al menos 1 semana entre ellas.
En particular, se necesita información sobre los efectos de la coadministración sobre las respuestas inmunitarias y la seguridad para formular una política de salud pública a la luz de los programas de vacunación simultáneos. Esta información es particularmente importante ya que la inmunosenescencia podría dejar a los adultos mayores más vulnerables a la infección, las complicaciones y la mortalidad por influenza, así como reducir sus respuestas inmunes a las vacunas estándar contra la influenza.4 La orientación actual en el Reino Unido es separar la administración de cualquier vacuna contra la COVID-19 y la influenza desplegada por al menos 7 días para evitar la atribución incorrecta de posibles eventos adversos.3 Los Centros para el Control de Enfermedades (CDC) de los Estados Unidos recomiendan un intervalo de 14 días entre estas vacunas.5 Sin embargo, la necesidad de múltiples visitas a la clínica podría conducir a un cumplimiento reducido y, por lo tanto, a una menor aceptación de la vacunación. Para garantizar una aceptación adecuada de las vacunas contra la COVID-19 y la influenza, la administración conjunta alentaría al público a tomar estas vacunas en una sola visita en lugar de regresar 7 días o más tarde.A continuación, se presentan los resultados de un subestudio de un ensayo de fase 3 en el Reino Unido que evaluó la seguridad y eficacia de dos dosis de NVX-CoV2373 en comparación con placebo.6 En el estudio principal, un total de 15 187 participantes se sometieron a aleatorización, de los cuales 15 139 participantes recibieron al menos una dosis de NVX-CoV2373 (n = 7569) o placebo (n = 7570), y 14 039 se incluyeron en la población de eficacia por protocolo. De la población de eficacia por protocolo, 3910 (27,9%) tenían 65 años o más, y 3117 (44,6%) tenían enfermedades coexistentes. Se observó una eficacia vacunal del 89,7% (IC del 95%: 80,2–94,6) contra la COVID-19 sintomática probada por PCR. La reactogenicidad fue generalmente leve y transitoria, y la incidencia de eventos adversos graves fue baja y similar en los dos grupos.6 En este subestudio, nuestro objetivo fue evaluar la seguridad, inmunogenicidad y eficacia de NVX-CoV2373 cuando se administra conjuntamente con una vacuna contra la influenza estacional autorizada.
Investigación en contexto
Evidencia antes de este estudio
Se realizaron búsquedas de artículos de investigación publicados en PubMed desde el 1 de diciembre de 2019 hasta el 1 de abril de 2021, sin restricciones de idioma mediante los términos «SARS-CoV-2», «COVID-19», «vacuna», «coadministración» e «inmunogenicidad». No se informaron publicaciones revisadas por pares que describieran el uso simultáneo de cualquier vacuna contra el SARS-CoV-2 y otra vacuna. Varios fabricantes de vacunas tenían publicaciones sobre los resultados de los ensayos de fase 3 de la vacuna contra el SARS-CoV-2 (Pfizer/BioNTech, Moderna, AstraZeneca, Janssen y el Instituto de Investigación gamaleya de Epidemiología y Microbiología). Ni estas publicaciones ni los protocolos de sus ensayos clínicos (cuando están disponibles públicamente) describían la coadministración, y a menudo tenían criterios de ensayo que excluían específicamente a aquellos con vacunación reciente o planificada con cualquier vacuna autorizada cerca o en el momento de cualquier inyección del estudio.
Valor añadido de este estudio
La interferencia inmune y la seguridad son siempre una preocupación cuando se administran dos vacunas al mismo tiempo. Hasta donde sabemos, este subestudio es el primero en mostrar el perfil de seguridad e inmunogenicidad y la eficacia clínica de la vacuna de una vacuna COVID-19 cuando se administra conjuntamente con una vacuna contra la influenza estacional.
Implicaciones de toda la evidencia disponible
Este estudio proporciona información muy necesaria para ayudar a guiar la toma de decisiones de la política nacional de inmunización sobre el importante tema del uso concomitante de las vacunas COVID-19 con las vacunas contra la influenza.
Resultados
Entre el 28 de septiembre de 2020 y el 28 de noviembre de 2020, un total de 15 187 participantes fueron asignados al azar al ensayo principal de fase 3; de los cuales 15 139 participantes recibieron al menos una dosis de NVX-CoV2373 (n = 7569) o placebo (n = 7570), de los cuales 431 fueron vacunados conjuntamente con una vacuna contra la influenza estacional (217 recibieron NVX-CoV2373 más la vacuna tetravalente basada en células de influenza o la vacuna trivalente contra la influenza adyuvante, según la edad, y 214 recibieron placebo más la vacuna tetravalente basada en células de influenza o la vacuna trivalente contra la influenza adyuvante; figura 1). Los que se inscribieron en el subestudio de influenza (n = 431) fueron retirados de la población de intención de tratar del estudio principal (n = 15 139), lo que resultó en la población de seguridad de intención de tratar del estudio principal (n = 14 708) utilizada para hacer comparaciones de seguridad con los participantes del subestudio de influenza. En el grupo de subestudio de influenza, 190 (43·3%) de 431 eran mujeres, 327 (75·1%) eran blancos, 98 (22·7%) eran de minorías étnicas o reportaron múltiples razas, y 117 (27·1%) tenían al menos una afección comórbida según las definiciones de los CDC de los Estados Unidos.5 La mediana de edad de los participantes del subestudio fue de 39 años, 142 (32,9%) de 431 tenían 50 años o más, y 29 (6,7%) tenían 65 años o más(apéndice p 12). Dentro del subestudio, 29 (6,7%) de 431 participantes recibieron la vacuna trivalente adyuvante contra la influenza con una mediana de edad de 66 años (n = 16) en el grupo NVX-CoV2373 y 69 años (n = 13) en el grupo placebo, y 402 (93, 3%) recibieron la vacuna tetravalente basada en células de influenza con una mediana de edad de 38 años (n = 201) en el grupo NVX-CoV2372 y 37 años (n = 201) en el grupo placebo (tabla 1).
Tabla 1Demografía y características basales de los participantes en el subestudio de administración conjunta de la vacuna contra la influenza y de las poblaciones completas del estudio
Los datos son media (DE), mediana (rango) o n (%). Los porcentajes se basan en el conjunto de datos itt dentro del subestudio de la vacuna contra la influenza estacional por tipo de vacuna (aTIV para aquellos ≥65 años y QIVc para aquellos <65 años de edad) y en general. aTIV=vacuna trivalente adyuvante contra la influenza. ITT=intención de tratar. QIVc=vacuna tetravalente basada en células de la influenza.* Los participantes con comorbilidades fueron aquellos identificados que tienen al menos una de las afecciones comórbidas informadas como antecedentes médicos o tienen un valor de índice de masa corporal de detección superior a 30 kg / m2.
Se evaluaron un total de 431 participantes para detectar eventos adversos no solicitados, eventos adversos graves, eventos adversos atendidos médicamente y eventos adversos de interés especial, y 404 participaron en la evaluación de la reactogenicidad. Los 431 participantes formaron parte de la población de inmunogenicidad evaluable tanto para el ensayo de inhibición de la hemaglutinación como para el ensayo igG de proteína anti-espiga. El grupo de subestudio en general fue más joven, más diverso racialmente y tuvo menos afecciones comórbidas que los participantes en el estudio principal, así como en las cohortes principales de reactogenicidad e inmunogenicidad del estudio (tabla 1; apéndice pp 12-13). La cohorte principal de inmunogenicidad del estudio para el ensayo IgG de proteína anti-espiga incluyó a 999 participantes en la población con intención de tratar que habían recibido la vacuna NVX-CoV2373 o placebo solo. La cohorte principal de reactogenicidad del estudio incluyó 2310 de la población de seguridad que había recibido al menos una dosis de la vacuna NVX-CoV2373 o placebo solo(figura 1).Overall, local reactogenicity (assessed only at the non-influenza vaccine injection site) was largely absent or mild in the co-administration group, NVX-CoV2373 alone group, and placebo plus influenza vaccine group (figure 2). Any local adverse event was reported in 122 (70·1%) of 174 co-vaccinated (three [1·7%] were severe), 640 (57·6%) of 1111 in the NVX-CoV2373 alone group (11 [1·0%] severe), 71 (39·4%) of 180 in the placebo plus influenza vaccine group (0% severe), and 195 (17·9%) of 1092 in the placebo alone group (two [0·2%] severe). The most commonly reported local adverse events were injection site tenderness (113 [64·9%] of 174 co-vaccinated and 592 [53·3%] of 1111 given NVX-CoV2373 alone) and injection site pain (69 [39·7%] co-vaccinated and 325 [29·3%] given NVX-CoV2373 alone).
Se informó cualquier evento adverso sistémico en 104 (60,1%) de 173 covacunados (cinco [2,9%] fueron graves), 506 (45,7%) de 1108 en el grupo nvX-CoV2373 solo (14 [1,3%] grave), 85 (47,2%) de 180 en el grupo de placebo más vacuna contra la influenza (cinco [2,8%] graves) y 397 (36,3%) de 1093 en el grupo de placebo solo (12 [1,1%] grave). En general, la incidencia de eventos específicos de reactogenicidad sistémica fue similar dentro de todos estos grupos (figura 2). Los eventos adversos sistémicos informados con mayor frecuencia fueron dolor muscular (49 [28,3%] de 173 covacunados y 237 [21,4%] de 1108 que recibieron NVX-CoV2373 solo) y fatiga (48 [27,7%] covacunados y 215 [19,4%] que recibieron NVX-CoV2373 solo), y el dolor muscular también ocurrió con más frecuencia en el grupo de administración conjunta que en el grupo de placebo más vacuna contra la influenza (49 [28,3%] de 173 vs 36 [20·0%] de 180). En particular, se notificó fiebre (es decir, temperatura ≥38 ° C) en siete (4· 3%) de 163 en el grupo covacunado, 21 (2· 0%) de 1067 en el grupo NVX-CoV2373 solo, tres (1· 7%) de 172 en el grupo de placebo más vacuna contra la influenza, y 16 (1· 5%) de 1061 en el grupo de placebo solo(apéndice pp 14-17).Cuando se evaluó por tipo específico de vacuna contra la influenza (es decir, la vacuna tetravalente basada en células de influenza en aquellos <65 años y la vacuna trivalente contra la influenza adyuvante en aquellos ≥65 años) entre las administradas concomitantemente con NVX-CoV2373, se observó una tendencia hacia tasas más bajas de reactogenicidad local y sistémica en el grupo de mayor edad que recibió la vacuna trivalente contra la influenza adyuvante. Cabe destacar que la mediana de duración de los eventos de reactogenicidad fue generalmente de 1 a 2 días para los eventos adversos locales y aproximadamente 1 día para los eventos adversos sistémicos tanto en el grupo covacunado como en el grupo NVX-CoV2373 solo; cuando se evaluó por tipo específico de vacuna contra la influenza, se observó una tendencia general para una duración más corta de la reactogenicidad entre las personas de 65 años o más (es decir, los receptores de la vacuna trivalente adyuvante contra la influenza; (datos no mostrados).Los eventos adversos no solicitados informados hasta 21 días después de la primera vacunación fueron predominantemente de gravedad leve y se distribuyeron de manera similar entre los grupos covacunados y NVX-CoV2373 solos(tabla 2). La frecuencia de todos los eventos adversos (40 [18,4%] de 217) y de todos los eventos adversos graves (uno [0,5%]) en el grupo covacunado fue similar a los del grupo NVX-CoV2373 solo (1297 [17,6%] de 7352 para todos los eventos adversos y 33 [0,4%] para todos los eventos adversos graves). Estas tasas también fueron similares a las tasas de todos los eventos adversos (31 [14,5%] de 214) y todos los eventos adversos graves (ninguno) en el grupo de placebo más vacuna contra la influenza y el grupo de placebo solo (1030 [14,0%] de 7356 para todos los eventos adversos y 33 [0,4%] para todos los eventos adversos graves; cuadro 2). Los eventos adversos no solicitados que ocurrieron en más del 1% del grupo covacunado incluyeron cefalea (cinco [2,3%] de 217), fatiga (cuatro [1,8%]) y dolor orofaríngeo (tres [1,4%]). El número de todos los eventos adversos atendidos médicamente fue de 17 (7,8%) de 217 en los vacunados conjuntamente y 279 (3,8%) de 7352 en los que recibieron NVX-CoV2373 solo, mientras que el número de eventos adversos atendidos médicamente fue de 18 (8,4%) de 214 en el grupo de placebo más vacuna contra la influenza y 288 (3,9%) de 7356 en el grupo placebo solo. El número de eventos adversos atendidos médicamente relacionados con el tratamiento fue menor y equilibrado en todos los grupos (tabla 2). El número de eventos adversos graves también fue bajo y equilibrado entre los participantes del subestudio y los que no participaron en el subestudio. No se informaron eventos adversos graves relacionados con el tratamiento en los participantes del subestudio. No se observaron afecciones médicas potencialmente inmunomediadas o eventos adversos de intereses especiales relevantes para COVID-19 en el subestudio de coadministración de influenza, con tasas de eventos resultantes similares a las que no participaron en el subestudio. No se informaron episodios de anafilaxia ni muertes dentro del subestudio.Tabla 2 Datosde seguridad de los participantes en el subestudio de administración conjunta de la vacuna contra la influenza y los participantes en toda la población del estudio por intención de tratar (sin participantes del subestudio)
NVX-CoV2373 más vacuna contra la influenza (n=217)
Placebo más vacuna contra la influenza (n=214)
NVX-CoV2372 solo (n=7352)
Placebo solo (n=7356)
Cualquier evento adverso
40 (18·4%)
31 (14·5%)
1297 (17·6%)
1030 (14·0%)
Cualquier evento adverso grave
1 (0·5%)
0
33 (0·4%)
33 (0·4%)
Evento adverso grave
1 (0·5%)
0
43 (0·6%)
44 (0·6%)
Evento adverso atendido médicamente
17 (7·8%)
18 (8·4%)
279 (3·8%)
288 (3·9%)
Evento adverso médicamente atendido relacionado con el tratamiento
3 (1·4%)
0
34 (0·5%)
17 (0·2%)
Condición médica potencialmente inmunomediada
0
0
5 (<0·1%)
8 (0·1%)
Evento adverso de especial interés relacionado con COVID-19
0
0
8 (0·1%)
22 (0·3%)
Los eventos adversos no solicitados y los eventos adversos graves son aquellos dentro de los 21 días posteriores a la dosis uno del estudio (con o sin administración conjunta de la vacuna contra la influenza). Los eventos adversos graves, los eventos adversos atendidos médicamente, los eventos adversos de interés especial y las afecciones médicas potencialmente inmunomediadas se evalúan durante todo el período de estudio.* La población principal de intención de tratar del estudio (n = 15 139) fueron todos los participantes que recibieron al menos una dosis de NVX-CoV2373 o placebo; los que se inscribieron en el subestudio de influenza (n = 431) se eliminaron para crear la población de seguridad principal del estudio (n = 14 708) para la comparación con los participantes del subestudio.
No se observaron diferencias significativas en los títulos medios geométricos basales de la inhibición de la hemaglutinación entre los del subgrupo covacunados con NVX-CoV2373 más el grupo de vacuna contra la influenza y los del grupo placebo más la vacuna contra la influenza(figura 3). En los grupos de vacunas tetravalentes basadas en células de la influenza, los títulos medios geométricos de inhibición de la hemaglutinación fueron significativamente más altos después de la vacunación en el día 21 en comparación con el día 0(apéndice pp 18-19). No se observaron diferencias en los títulos medios geométricos del día 21 de la inhibición de la hemaglutinación entre el grupo de vacuna contra la influenza NVX-CoV2373 más y el grupo de vacuna placebo más influenza para cualquier cepa de influenza individual (A/H1N1, A/H3N2, B/Victoria o B/Yamagata) para cualquiera de las vacunas contra la influenza. Los valores de aumento medio geométrico del pliegue siguieron el mismo patrón(apéndice pp 18-19). Tanto para la vacuna tetravalente basada en células de influenza como para la vacuna trivalente adyuvante contra la influenza, las tasas de seroconversión de inhibición de la hemaglutinación fueron generalmente más altas para las cepas de influenza A que para las cepas de influenza B (figura 4).
Las unidades de ELISA IgG de proteína antipijo basal fueron similares en los participantes en el subestudio covacunados con NVX-CoV2373 más la vacuna contra la influenza y en los que recibieron placebo más la vacuna contra la influenza, así como en los vacunados en la cohorte de inmunogenicidad del estudio principal con NVX-CoV2373 solo o placebo solo (los datos para la población de inmunogenicidad por protocolo se muestran en la tabla 3 ). En los grupos vacunados con NVX-CoV2373 más la vacuna contra la influenza o con NVX-CoV2373 solo, las unidades ELISA medias geométricas del día 35 fueron significativamente más altas que las del inicio. Se observó una diferencia en las unidades ELISA medias geométricas entre los dos grupos por protocolo (31 236·1 [IC del 95%: 26 295–5–37 104·9] para el grupo nvX-CoV2373 más la vacuna contra la influenza [n=178] vs 44 678·3 [40 352·2–49 468·2] para el grupo NVX-CoV2373 solo [n=414]). Una evaluación post-hoc de la relación entre las dos medias geométricas cuando se ajustó por unidades ELISA basales, edad y grupo de tratamiento fue de 0,57 (IC del 95%: 0,47–0,70). Esta diferencia entre la cohorte de la vacuna contra la influenza NVX-CoV2373 más y la cohorte NVX-CoV2373 sola también se reflejó en los aumentos de pliegues medios geométricos, pero no en las tasas de seroconversión. La media geométrica del día 35 en el grupo de vacunación concomitante elitista fue numéricamente menor en los de 65 años o más (es decir, los que recibieron la vacuna trivalente contra la influenza adyuvante) en comparación con los de 18 a menos de 65 años (es decir, los que recibieron la vacuna tetravalente basada en células de influenza) grupo de vacunación concomitante, aunque el número de participantes en el grupo concomitante de vacunación contra la influenza trivalente adyuvante fue pequeño. Sin embargo, los aumentos de pliegue medio geométrico en ambos grupos fueron grandes con más de 200 veces en comparación con el día 0, y las tasas de seroconversión fueron más del 97%. Esta disminución en la inmunogenicidad con el aumento de la edad también se observó en la cohorte principal de inmunogenicidad del estudio. El subgrupo de participantes que recibieron NVX-CoV2373 concomitante y cualquier vacuna contra la influenza que fueron seropositivos (n = 19) al inicio del estudio lograron unidades ELISA medias geométricas del día 35 que fueron significativamente mayores que las de participantes similares que fueron seronegativas (n = 198) al inicio del estudio (71 115·6 [IC del 95%: 46 813·0–108 032·8] vs 30 439·1 [25 713·4–36 033·5]; apéndice p 20).Tabla 3Proteína antipigada IgG en el día 0 y el día 35 en el subestudio de vacunación antigripal y en la cohorte de inmunogenicidad de la población por protocolo
NVX-CoV2373 más vacuna contra la influenza
Placebo más vacuna contra la influenza
NVX-CoV2373 solo
Placebo solo
n
Día 0
Día 35
n
Día 0
Día 35
n
Día 0
Día 35
n
Día 0
Día 35
Media geométrica ELISA unit
Vacuna inactivada contra la influenza más NVX-CoV2372 o placebo (todas las edades)
n=178
116·3 (107·7–125·6)
31 236·1 (26 295·5–37 104·9)
n=181
111·4 (105·1–118·1)
115·7 (106·1–126·1)
n=414
112·2 (107·5–117·0)
44 678·3 (40 352·2–49 468·2)
n=417
110·3 (106·3–114·5)
113·2 (106·8–120·0)
QIVc más NVX-CoV2373 o placebo (18 a <65 años)
n=168
115·8 (107·2–125·0)
31 516·9 (26 316·2–37 745·3)
n=170
112·2 (105·4–119·3)
116·8 (106·5–128·0)
n=300
111·9 (106·2–117·9)
47 564·3 (42 327·3–53 449·4)
n=310
109·7 (105·2–114·4)
113·5 (105·6–122·0)
aTIV más NVX-CoV2373 o placebo (≥65 años)
n=10
125·6 (75·0–210·3)
26 876·1 (15 374·6–46 981·5)
n=11
100·0 (100·0–100·0)
100·0 (100·0–100·0)
n=114
112·8 (105·0–121·2)
37 892·8 (30 833·3–46 568·5)
n=107
112·1 (103·4–121·4)
112·3 (103·1–122·3)
Aumento geométrico del pliegue medio
Vacuna inactivada contra la influenza más NVX-CoV2372 o placebo (todas las edades)
n=178
NA
268·6 (221·0–326·4)
n=181
NA
1·0 (1·0–1·1)
n=414
NA
398·4 (358·6–442·6)
n=417
NA
1·0 (1·0–1·1)
QIVc más NVX-CoV2373 o placebo (18 a <65 años)
n=168
NA
272·3 (222·3–333·5)
n=170
NA
1·0 (1·0–1·1)
n=300
NA
425·0 (375·7–480·8)
n=310
NA
1·0 (1·0–1·1)
aTIV más NVX-CoV2373 o placebo (≥65 años)
n=10
NA
214·0 (96·5–474·6)
n=11
NA
1·0 (1·0–1·0)
n=114
NA
335·9 (274·4–411·1)
n=107
NA
1·0 (1·0–1·0)
Tasa de seroconversión
Vacuna inactivada contra la influenza más NVX-CoV2372 o placebo (todas las edades)
n=178
NA
97·8 (94·3–99·4)
n=181
NA
0·6 (0·0–3·0)
n=414
NA
99·0 (97·5–99·7)
n=417
NA
0·7 (0·1–2·1)
QIVc más NVX-CoV2373 o placebo (18 a <65 años)
n=168
NA
97·6 (94·0–99·3)
n=170
NA
0·6 (0·0–3·2)
n=300
NA
99·0 (97·1–99·8)
n=310
NA
1·0 (0·2–2·8)
aTIV más NVX-CoV2373 o placebo (≥65 años)
n=10
NA
100·0 (69·2–100·0)
n=11
NA
0·0 (0·0–28·5)
n=114
NA
99·1 (95·2–100·0)
n=107
NA
0·0 (0·0–3·4)
Los datos son n o resultado del ensayo (IC del 95%). La vacuna inactivada contra la influenza incluyó tanto aTIV como QIVc. Los participantes del subestudio de administración conjunta de la vacuna contra la influenza se compararon con la población de inmunogenicidad por protocolo (se muestran datos de los participantes que consintieron en que se evaluaran las concentraciones de IgG). Comparación de las unidades ELISA medias geométricas de la proteína IgG antipiiga al inicio (día 0) y día 35, así como el aumento medio geométrico del pliegue del día 35 y la tasa de seroconversión después de la vacunación con NVX-CoV2373 o placebo con aTIV, QIVc o solo. aTIV=vacuna trivalente adyuvante contra la influenza. NA=no aplicable. QIVc=vacuna tetravalente basada en células de la influenza.
Entre los 386 participantes en el subestudio de influenza que se incluyeron en la población de eficacia por protocolo, se informó que dos (1%) de 191 participantes tenían COVID-19 sintomático confirmado virológicamente con inicio al menos 7 días después de la segunda dosis entre los receptores de la vacuna y ocho (4%) de 195 entre los receptores de placebo. Un análisis post-hoc del criterio de valoración primario mostró una eficacia de la vacuna del 74,8% (IC del 95%: -19,7 a 94,7) en el grupo de subestudio de influenza. Entre 360 participantes que tenían entre 18 y menos de 65 años, se informó que uno (<1%) de 178 participantes tenía COVID-19 sintomático confirmado virológicamente con inicio al menos 7 días después de la segunda dosis entre los receptores de la vacuna y ocho (4%) de 182 entre los receptores de placebo; eficacia de la vacuna del 87,5% (IC del 95%: -0,2 a 98,4; apéndice p 22). Se notificaron muy pocos casos entre aquellos en la población por protocolo que tenían 65 años o más para calcular la eficacia de una vacuna. Todos los casos de COVID-19 en los subestudios de influenza en el grupo por protocolo se debieron a la variante Alfa (B.1.1.7). Entre los 431 participantes en la población de intención de tratar del subestudio sobre la influenza, la eficacia de la vacuna fue del 80,6% (IC del 95%: 13,3–95,7; apéndice p 22). La eficacia de la vacuna en la población principal del estudio por protocolo (aquellos de 18 a <65 años) fue del 89,8% (IC del 95%: 79,7–95,5), mientras que la eficacia de la vacuna contra la variante Alfa sola en la población principal del estudio por protocolo fue del 86,3% (71,3–93,5).
Discusión
Hasta donde sabemos, este subestudio es el primero en mostrar la seguridad, inmunogenicidad y eficacia de cualquier vacuna COVID-19 cuando se administra conjuntamente con una vacuna contra la influenza estacional o cualquier otra vacuna. La mayoría de los ensayos de vacunas contra la COVID-19 han excluido a los participantes que reciben otras vacunas en el momento o cerca del momento de la inyección con la vacuna del estudio y, por lo tanto, no tienen estudios de interacción abordados en sus etiquetas.9, 10, 11
Aunque no se especificaron criterios de valoración de inmunogenicidad comparativa específicos en este subestudio exploratorio, no se encontró evidencia de interferencia de la vacuna contra la COVID-19 con la vacuna tetravalente basada en células de la influenza. Las conclusiones definitivas sobre la vacuna trivalente adyuvante contra la influenza no fueron posibles debido al pequeño número de participantes de 65 años o más. Sin embargo, observamos un efecto de la administración concomitante de una vacuna contra la influenza sobre la magnitud absoluta de la respuesta de anticuerpos IgG de la proteína anti-espiga. Este efecto no pareció ser clínicamente significativo, ya que la eficacia de la vacuna parecía preservarse. La administración conjunta tampoco pareció tener un efecto clínicamente significativo sobre la reactogenicidad sistémica o local y no se encontraron problemas de seguridad adicionales asociados con la covacunación.
Los eventos de reactogenicidad local y sistémica solicitados después de la coadministración fueron generalmente similares a la incidencia y gravedad de los de cada vacuna cuando se administraron por separado.
La incidencia de reactogenicidad local más subjetiva (es decir, dolor y sensibilidad) fue elevada en el grupo covacunado por encima del nivel de NVX-CoV2373 solo o placebo más los grupos de vacuna contra la influenza, pero las incidencias de eventos locales más objetivos (es decir, eritema e hinchazón) fueron bajas e indistinguibles entre todos los grupos. Este aumento de la incidencia fue impulsado en gran medida por un aumento en los síntomas leves. No está claro si los participantes estaban sesgados en su evaluación del dolor y la sensibilidad en el lugar de la inyección del estudio después de haber recibido dos vacunas coadministradas; el hecho de que se evaluara que las inyecciones de placebo causaban más dolor o sensibilidad local cuando se administraban concomitantemente con una vacuna contra la influenza (administrada en el brazo opuesto) en comparación con las inyecciones de placebo, cuando se administraban solas, sugeriría que es probable que este sea el caso. Otra explicación es que los participantes registraron los síntomas locales aditivos del sitio de inyección de influenza por error a pesar de que se les indicó que registraran los síntomas solo en el sitio de inyección de la vacuna del estudio. La incidencia de cualquier evento de reactogenicidad sistémica en los vacunados conjuntos fue modestamente elevada sobre la incidencia de NVX-CoV2373 o la vacuna contra la influenza sola, lo que concuerda con una mayor carga general de inmunógenos de la vacuna y la población participante relativamente más joven en el subestudio. Este efecto se observó principalmente para los eventos de dolor muscular y fiebre, sin embargo, a pesar del aumento relativo en la incidencia de fiebre, la tasa absoluta de fiebre en aquellos que recibieron dos vacunas coadministradas fue modesta (4· 3%). La incidencia de eventos graves fue baja en todos los grupos y no mostró un patrón clínicamente significativo de aumento de la reactogenicidad. La elevación en algunos eventos de reactogenicidad podría, en parte, haberse debido a la edad general más joven de los participantes del subestudio de la vacuna contra la influenza en comparación con la cohorte principal de reactogenicidad del estudio (mediana de edad de 39,0 años [93,3% de 18 a <65 años] frente a 52,0 años [80,1% de 18 a <65 años]). Aquellos que tenían 65 años o más que recibieron dos vacunas adyuvantes (es decir, la NVX-CoV2373 adyuvada y la vacuna trivalente adyuvante contra la influenza) en comparación con los menores de 65 años que recibieron una vacuna adyuvante (es decir, la NVX-CoV2373 adyuvada y la vacuna tetravalente sin adyuvante basada en células de influenza) tuvieron tasas más bajas de reactogenicidad; Este efecto de la edad también se observó en el grupo NVX-CoV2373 solo y en estudios anteriores de NVX-CoV23736, 12, 13 y es consistente con inmunosenescencia.La incidencia de eventos adversos, eventos adversos graves y eventos adversos de especial interés fue baja y equilibrada entre los que recibieron NVX-CoV2373, la vacuna contra la influenza o ambos. La incidencia de cualquier evento adverso atendido médicamente fue mayor en los participantes del subestudio en comparación con los participantes no subestudios. Esta diferencia fue menos evidente cuando se evaluaron los eventos adversos relacionados con el tratamiento atendidos médicamente solamente. El aumento de la tasa de todos los eventos adversos atendidos médicamente en el subestudio podría representar un sesgo de búsqueda de atención médica en aquellos que desean una vacuna contra la influenza en lugar de un verdadero aumento en las visitas médicas debido a eventos adversos relacionados con la vacunación conjunta o la recepción de la vacuna contra la influenza más placebo; una evaluación de estas visitas médicas excesivas reveló que la mayoría eran visitas de práctica general asociadas con problemas de mantenimiento de la salud (datos no mostrados).La magnitud de la respuesta humoral a ninguna de las vacunas contra la influenza no se vio afectada por la administración conjunta con NVX-CoV2372 cuando se evaluó a los 21 días después de la dosificación, aunque se debe tener cuidado al generalizar esta observación a la vacuna trivalente contra la influenza adyuvante debido al pequeño tamaño de la muestra. El aumento posterior a la vacunación en los títulos medios geométricos y las tasas de seroconversión para cada cepa fueron altos cuando se administró la vacuna contra la influenza con placebo o NVX-CoV2373, aunque se observó una respuesta generalmente menor a las cepas de influenza B en todos los receptores de la vacuna contra la influenza. La respuesta inmune humoral a las cepas de influenza B depende de numerosos factores, incluida la edad y la exposición previa a la vacuna contra la influenza.14
Bajas tasas de seroconversión de la influenza B15 y tasas de seroconversión más bajas en relación con las cepas de influenza A16, 17 se han observado con estudios previos de inmunogenicidad de vacunas tetravalentes inactivadas contra la influenza.Por el contrario, se observó una reducción modesta en las unidades elISA igG de proteína antipigada con la coadministración de NVX-CoV2373 y una vacuna contra la influenza. No está claro si esta reducción se debió a la interferencia de la vacuna o a la naturaleza no aleatoria de los grupos estudiados.
En ausencia de un correlato de protección, la interpretación de la importancia de esta constatación es difícil.
La evaluación post-hoc de la eficacia de la vacuna en este subestudio en personas de 18 a menos de 65 años fue del 87,5% en comparación con la eficacia de la vacuna del 89,8% en el mismo grupo de edad de las poblaciones de eficacia por protocolo en el estudio principal.
La eficacia similar de la vacuna para el grupo de subestudio de coadministración y el grupo de estudio principal sugeriría que la reducción en las unidades ELISA de proteína IgG anti-espiga como resultado de la coadministración podría no ser clínicamente significativa. De hecho, las concentraciones de unidades ELISA IgG de proteína anti-espiga en aquellos que recibieron ambas vacunas (ya sea en aquellos de 18 a <65 años o ≥65 años) fueron aún más de tres veces mayores que las unidades de PROTEÍNA IGG ELISA anti-espiga que se encuentran en el suero convaleciente, lo que sugiere que las unidades ELISA en este rango encontradas en los participantes del subestudio podrían ser protectoras.18, 19
También debe tenerse en cuenta que no se observaron diferencias en las tasas de seroconversión entre los vacunados conjuntamente y los que recibieron NVX-CoV2373 solo.
El alcance de la reducción en las unidades ELISA igG de proteína antipigo podría ser menos relevante en los participantes que son seropositivos al inicio del estudio, ya que alcanzaron valores altos después de la vacunación con la administración conjunta de la vacuna contra la influenza con una media de 71 115 unidades ELISA en participantes seropositivos vacunados de todas las edades en comparación con una media de 46 679 unidades ELISA en los receptores NVX-CoV2373 solos por protocolo de todas las edades (sin embargo, este hallazgo no fue tan grande como la media de 125 490 unidades ELISA en receptores seropositivos de NVX-CoV2372 solos). Una posible explicación para este hallazgo es que los individuos seropositivos tienen poblaciones preexistentes de células T y células B con memoria inmune contra la proteína espiga del SARS-CoV2, minimizando cualquier posible efecto de interferencia inmune. Por lo tanto, la administración conjunta de la vacuna contra la influenza podría afectar el cebado, pero no tener ningún efecto sobre la respuesta inmune en individuos previamente preparados.
Una implicancia de este hallazgo es que la administración conjunta de la vacuna contra la influenza con la segunda dosis de cualquier programa de vacunas contra el COVID-19 de dos dosis, o con una dosis de refuerzo posterior de la vacuna contra el COVID-19, podría superar cualquier posible interferencia inmunitaria.
Este efecto debe evaluarse más a fondo, ya que tiene implicaciones importantes para las estrategias de vacunación de salud pública.
Aunque este subestudio es el primero en evaluar la administración conjunta de una vacuna contra la COVID-19 con una vacuna contra la influenza estacional, la administración conjunta de la vacuna contra la influenza en otros entornos ha sido bien estudiada.
Nuestro estudio utilizó dos vacunas diferentes contra la influenza para diferentes grupos de edad de conformidad con las pautas de vacunación contra la influenza del Reino Unido.3
Para los menores de 65 años, se utilizó una vacuna tetravalente inactivada contra la influenza derivada de cultivos celulares. La vacuna tetravalente basada en células de la influenza se aprobó en el Reino Unido en diciembre de 2018 para personas de 9 años o más y se extendió a 2 años o más en 2020.
Para la cohorte de mayor edad, se administró una vacuna trivalente contra la influenza trivalente a base de escualeno MF59, adyuvada con aceite en agua.
Esta vacuna adyuvante trivalente contra la influenza fue aprobada en el Reino Unido en agosto de 2017.
En dos estudios de la vacuna contra la influenza trivalente adyuvante MF59 administrada concomitantemente con una vacuna antineumocócica, las respuestas de anticuerpos a ninguna de las vacunas se vieron afectadas, y los datos de seguridad fueron consistentes con las tasas esperadas de eventos adversos para ambas vacunas.20, 21
No se han reportado problemas de interferencia o seguridad con una vacuna tetravalente contra la influenza administrada conjuntamente con vacunas antineumocócicas y herpes zóster.22, 23
Las fortalezas de nuestro subestudio incluyen el diseño del estudio controlado con placebo y su alineación con la política nacional de vacunas contra la influenza del Reino Unido en el uso de vacunas contra la influenza adyuvantes y no adyuvantes en diferentes grupos de edad. Las limitaciones de los estudios incluyen el pequeño tamaño general del subestudio (con pocos participantes ≥65 años debido a la alta tasa de vacunación sistemática contra la influenza entre los participantes de este grupo de edad al inicio del estudio), el pequeño número de criterios de valoración de eficacia del subestudio, la ausencia de una evaluación estadística formal preespecificada de la inmunogenicidad y la ausencia de asignación al azar en el reclutamiento del subestudio de influenza. cohortes de inmunogenicidad y reactogenicidad.
Un diseño de estudio más sólido podría haber involucrado a cuatro grupos aleatorios, que consisten en NVX-CoV2373 más vacuna contra la influenza, NVX-CoV2373 más placebo, vacuna contra la influenza más placebo y placebo más placebo.
Otra limitación fue el diseño abierto del estudio en la administración de la vacuna contra la influenza, pero este diseño fue necesario para permitir a los participantes considerar solo el sitio de inyección de la vacuna del estudio para la evaluación de los síntomas locales.
Finalmente, la evaluación de los títulos de anticuerpos neutralizantes podría haber beneficiado la investigación de inmunogenicidad, sin embargo, estudios previos con NVX-CoV2373 han demostrado una fuerte correlación entre la proteína anti-espiga y los resultados de microneutralización de tipo salvaje.18
En conclusión, este subestudio es el primero en mostrar el perfil de seguridad, inmunogenicidad y eficacia de una vacuna contra el COVID-19 cuando se administra conjuntamente con una vacuna contra la influenza estacional.
Estos datos no muestran problemas de seguridad tempranos con la administración concomitante de NVX-CoV2373 con una vacuna contra la influenza.
La inmunogenicidad de la vacuna contra la influenza se preservó con la administración concomitante, aunque se encontró una disminución modesta en la inmunogenicidad de la vacuna NVX-CoV2373. La eficacia de la vacuna en las personas de 18 a menos de 65 años pareció preservarse en las personas que recibieron ambas vacunas en comparación con las vacunadas con NVX-CoV2373 sola.
Los ensayos clínicos futuros y los estudios posteriores a la licencia de las vacunas contra la COVID-19 deben incluir datos de seguridad e inmunogenicidad para la coadministración con vacunas comunes para adultos y pediátricas.
Se necesita más investigación sobre la vacunación concomitante de las vacunas contra la COVID-19 y la gripe, especialmente en los mayores de 65 años, para ayudar a orientar la política nacional de inmunización sobre este importante tema.
Se transcriben varias evaluaciones, podría ser una política pública para acceder a otra estrategia de contención de la propagación de la pandemia, mejorar el aislamiento y evitar o mitigar la onda de propagación. Debemos estar alertas que caminos alternativos explorar, cómo se puede implementar una mejora en la equidad, en el acceso, en mitigar efectos pandémicos.
Es evidente que las vacunas en uso son fundamentales para la contención de la pandemia, disminuir casos graves y mortalidad, pero su efectividad disminuye muy rápidamente, especialmente para las mutaciones. Entiendo que debiera la ciencia, desarrollar otras tecnologías vacunales más efectivas, estrategias no solo de prevención de enfermedad grave y mortal, sino la transmisión, porque en caso contrario la cantidad de casos, con sus aislamientos respectivos llevará a una disminución de la fuerza productiva de trabajadores esenciales, de la producción de bienes, el intercambio y la reposición de algunos hábitos que incremente el crecimiento económico
Evaluación de la viabilidad de la autoprueba de antígeno de Biosynex COVID-19 AG + para la detección de la proteína nucleocápsida del SARS-CoV-2 a partir de las secreciones nasales del cornete medio autocolectadas en el público en general en Francia
Debido a su facilidad de uso, las pruebas de diagnóstico rápido de detección de antígeno SARS-CoV-2 de ensayo de flujo lateral podrían ser candidatas adecuadas para la autoprueba de diagnóstico rápido de detección de antígeno (Ag-RDST). Evaluamos la viabilidad de la autoprueba de antígeno Ag-RDST BIOSYNEX COVID-19 Ag + (Biosynex Swiss SA, Freiburg, Suiza), utilizando secreciones nasales recolectadas por ellos mismos del medio de cornetes (NMT), en 106 voluntarios adultos incluidos de manera prospectiva que viven en París, Francia. La mayoría de los participantes entendieron correctamente las instrucciones de uso (94,4%; intervalo de confianza (IC) del 95%: 88,3-97,4), mostrando una gran capacidad para realizar todo el procedimiento de autocomprobación para obtener un resultado válido e interpretable (100% ; IC del 95%: 96,5-100), y demostró la capacidad de interpretar correctamente los resultados de las pruebas (96,2%; IC del 95%: 94,2-97,5) con un alto nivel de satisfacción general. Aproximadamente uno de cada ocho participantes (# 15%) necesitó ayuda verbal para realizar o interpretar la prueba, y solo el 3.8% de los resultados de la prueba fueron mal interpretados. En referencia a la RT-PCR multiplex en tiempo real, el Ag-RDST mostró 90,9% y 100% de sensibilidad y especificidad, respectivamente, y un alto acuerdo (98,1%), fiabilidad (0,94) y precisión (90,9%) para detectar el SRAS. Antígeno CoV-2. Tomados en conjunto, nuestro estudio demuestra la alta usabilidad y precisión de BIOSYNEX Antigen Self-Test COVID-19 Ag + para el muestreo de NMT auto-recopilado supervisado en una población adulta no seleccionada que vive en Francia. y alta concordancia (98,1%), fiabilidad (0,94) y precisión (90,9%) para detectar el antígeno del SARS-CoV-2. Tomados en conjunto, nuestro estudio demuestra la alta usabilidad y precisión de BIOSYNEX Antigen Self-Test COVID-19 Ag + para el muestreo de NMT auto-recopilado supervisado en una población adulta no seleccionada que vive en Francia. y alta concordancia (98,1%), fiabilidad (0,94) y precisión (90,9%) para detectar el antígeno del SARS-CoV-2. Tomados en conjunto, nuestro estudio demuestra la alta usabilidad y precisión de BIOSYNEX Antigen Self-Test COVID-19 Ag + para el muestreo de NMT auto-recopilado supervisado en una población adulta no seleccionada que vive en Francia.
Si bien las pruebas de amplificación de ácido nucleico recomendadas actualmente, como los ensayos de reacción en cadena de la polimerasa con transcripción inversa en tiempo real, siguen siendo la piedra angular del estándar de oro para el diagnóstico de la infección por SARS-CoV-2 [ 4 , 5 ], los métodos inmunológicos también se pueden utilizar para detectar antígenos virales [ 5 , 6 , 7 ]. Por lo tanto, las pruebas de diagnóstico rápido de detección de antígenos (Ag-RDT) permiten nuevas estrategias de prueba para el diagnóstico y control de la infección por SARS-CoV-2 debido a su breve tiempo de respuesta y su facilidad de uso [ 6 , 8 , 9]. Sin embargo, la mayoría de las Ag-RDT de SARS-CoV-2 dependen del muestreo nasofaríngeo, y su uso se ve obstaculizado por la necesidad de desplegar trabajadores sanitarios cualificados y equipo de protección para recoger las secreciones nasofaríngeas, realizar las pruebas e interpretar los resultados de las mismas. Además, un hisopado nasofaríngeo a menudo se considera incómodo, lo que limita la capacidad de realizar con frecuencia pruebas de detección de este tipo de muestreo nasofaríngeo [ 10 ].Los métodos de muestreo alternativos, como el muestreo nasal del cornete medio (NMT) (se inserta un hisopo unos 2-3 cm en la fosa nasal paralela al paladar hasta que se encuentre resistencia en los cornetes), son bien tolerados y pueden ser realizados por la propia persona sin supervisión a domicilio o supervisado in situ [ 10 , 11 , 12 , 13 ]. El muestreo de NMT fue equivalente al muestreo nasofaríngeo para un SARS-CoV-2 Ag-RDT incluido en la lista de la OMS, que sienta las bases para su uso como una técnica de auto-muestreo bajo supervisión [ 12 , 13 , 14 , 15 ]. El uso generalizado de autopruebas para la infección por SARS-CoV-2 podría ayudar a mejorar el control de la propagación de la epidemia de SARS-CoV-2 [ 16, 17 , 18 , 19 , 20 , 21 ]. La experiencia previa con la autoprueba del VIH mostró que una autoprueba la puede realizar perfectamente una persona común y da resultados fiables y precisos, y que la autoprueba del VIH se considera una herramienta aceptable y útil para mejorar la cobertura de la prueba del VIH en todo el mundo para mejorar Control de la epidemia del VIH [ 22 , 23 ]. Las pruebas de flujo lateral para el antígeno del SARS-CoV-2 son apropiadas para su uso como una autoprueba de diagnóstico rápido de detección de antígeno (Ag-RDST) debido a su bajo costo, facilidad de uso y resultado rápido [ 12 , 14 , 24 ] .Informes recientes han establecido la viabilidad del automuestreo de NMT bajo supervisión [ 12 , 14 ]. Sin embargo, los datos sobre el rendimiento clínico de la autocomprobación con Ag-RDT se limitan a comparaciones con la detección cuantitativa de RT-PCR en tiempo real (rtRT-PCR) del ARN del SARS-CoV-2 [ 25 , 26 ].Este estudio tuvo como objetivo evaluar la usabilidad y el rendimiento clínico en el campo de la autoprueba de antígeno COVID-19 Ag + de Ag-RDST BIOSYNEX (Biosynex Swiss SA, Freiburg, Suiza; referencia 859271) para las pruebas antigénicas de COVID-19 de NMT recolectado por ellos mismos. secreciones en adultos que viven en la región de París durante la tercera ola de la epidemia de COVID-19 en Francia. Por primera vez que sepamos, la usabilidad de este novedoso Ag-RDST se evaluó de acuerdo con un enfoque sistemático basado en nuestra experiencia previa en la evaluación de la autoprueba del VIH y el SARS-CoV-2 utilizando muestras de sangre capilar [ 27 , 28 , 29 , 30 ].
Comparativa de los test disponibles para coronavirus
Molecular (prueba de PCR)
Antígeno
Anticuerpo (Inmunocromatografía o test rápido y ELISA /CLIA)
Tipo de muestra
Nasofaríngeo, nasal o garganta.
Nasofaríngeo o nasal.
Sangre capilar para inmunocromatografía y suero o plasma para ELISA/CLIA.
Resultado que muestra
Infección activa (detecta la presencia de genoma del virus).
Infección activa (detecta la presencia de proteínas del virus).
Infección pasada o en fases finales (presencia de anticuerpos IgM e IgG frente al virus).
Situaciones en las que se recomienda su uso
Patrón de referencia para el diagnóstico de pacientes con síntomas o contacto estrecho en los primeros 15 días desde el inicio de síntomas o contacto (desde unos días antes de la aparición de los síntomas hasta el final de la segunda semana).
Pacientes con síntomas o contacto estrecho durante los 5 primeros días desde el inicio de síntomas o contacto y alta carga viral, si no se puede realizar una PCR, o se necesitan resultados rápidos para el manejo clínico.
IgM (6-20 días) IgG (a partir del día 15) desde el inicio de síntomas o contacto. Estudios de seroprevalencia (población que ha estado en contacto con el virus); Evaluación clínica de personas con enfermedad tardía y de manera conjunta con PCR; Sospecha de síndrome postinfeccioso.
Sensibilidad (en situaciones en las que se recomienda su uso)
Alta
Moderada
Moderada (Inmunocromatografía) y Alta (ELISA/CLIA)
Especificidad (en situaciones en las que se recomienda su uso)
Alta
Alta
Alta (aunque menor en inmunocromatografía)
Situaciones en las que se recomienda su uso y puede dar un resultado falso positivo
Detección de restos de genoma viral, no infeccioso.
Reacción cruzada con otros virus.
Situaciones en las que se recomienda su uso y puede dar un resultado falso negativo
Escasa eliminación del virus (estadío clínico, gravedad).
Escasa eliminación del virus (estadío clínico, gravedad). Si hay sospecha de infección, confirmar con PCR.
Fase precoz de la infección.
Tabla: Blanca Lumbreras (UMH) y Salvador Peiró (Fisabio) Descargar los datos
Población de estudio
Se evaluó la elegibilidad de un total de 116 personas, pero se excluyó a 10 porque eran menores de 18 años (n = 4) y no daban su consentimiento ( n = 6) ( Figura 1 ). Finalmente, 106 se inscribieron con éxito en todos los subestudios de viabilidad y evaluación del desempeño clínico.Las características demográficas y el historial médico de los participantes del estudio se muestran en la Tabla 1 . En total, 68 (64,2%) eran mujeres. La mediana de edad fue de 40 años y la mayoría de los participantes tenían entre 21 y 59 años. La mayoría (83,9%) de los participantes poseía al menos un nivel universitario o de educación superior. La mayoría (87,7%) de ellos no poseía experiencia previa en la autoevaluación de enfermedades distintas de COVID-19.3.1. Las principales razones para realizar las pruebas fueron los viajes en avión ( n = 41; 38,7%), la exposición de un caso por contacto de un individuo infectado con SARS-CoV-2 ( n = 10; 9,4%), sospecha de COVID-19 ( n = 30; 28,3%) ), evaluación preoperatoria ( n = 18; 17,0%) y control de la infección por SARS-CoV-2 en los 30 días anteriores (n = 7; 6,6%). En el momento de la prueba, la mayoría (71,7%) de los participantes no habían informado síntomas de COVID-19 en el último mes, incluidos todos los casos de contacto de personas infectadas con SARS-CoV-2. Alrededor de un tercio de los participantes informaron al menos un síntoma compatible con COVID-19. Entre los pacientes sintomáticos, el tiempo medio de duración de los síntomas antes de la toma de muestras fue de 4 días (rango, 0 a 7 días).
Discusión
En este documento informamos sobre nuestra experiencia reciente durante el tercer período pico de la epidemia de COVID-19 en Francia de la usabilidad y el rendimiento clínico en el campo de un nuevo test de antígenos rápidos y autoadministrados Ag-RDST para las pruebas antigénicas de COVID-19 de las secreciones de NMT recolectadas por ellos mismos en voluntarios adultos que viven en la región de París. La mayoría de los participantes pudo comprender correctamente las instrucciones de uso de la autoevaluación, obtener un resultado fiable e interpretar los resultados finales de la prueba. Aproximadamente uno de cada ocho (# 15%) participantes necesitó ayuda verbal, y solo el 3,8% de los resultados de las pruebas se malinterpretaron. Tomados en conjunto, nuestro estudio demuestra la viabilidad y precisión de la autocomprobación de COVID-19 a partir de muestras de NMT recopiladas por ellos mismos en una población adulta no seleccionada en un entorno supervisado.
4.1. Evaluación de viabilidad
La usabilidad global del BIOSYNEX Antigen Self-Test COVID-19 Ag + fue evaluada mediante cuatro subestudios sucesivos que permiten un buen análisis de todos los pasos de la realización de la prueba, desde la comprensión de las instrucciones de uso con la identificación de la prueba. componentes, hasta la realización del Ag-RDST y la interpretación final de los resultados. El subestudio de satisfacción también es fundamental para evaluar la aceptabilidad de la autocomprobación, que necesariamente deberá repetirse en un contexto epidémico.El subestudio 1 evaluó si todos los participantes podían leer y comprender las instrucciones de uso. Nuestros hallazgos mostraron que el 94,4% de los participantes respondieron correctamente las 11 preguntas, lo que indica una comprensión generalmente correcta de los mensajes clave entregados por las instrucciones de uso de BIOSYNEX Antigen Self-Test COVID-19 Ag +, con una tasa general de buenas respuestas de 96,8 %. Estos resultados satisfactorios pueden explicarse en parte por un nivel de educación suficiente de la mayoría de los participantes del estudio. De hecho, la experiencia previa con el autodiagnóstico del VIH ha demostrado que un nivel de educación insuficiente es un gran desafío para comprender las instrucciones de uso [ 22 , 40]. Aunque las revisiones sistemáticas y el metanálisis han demostrado que el autodiagnóstico del VIH puede ser realizado con éxito por usuarios no capacitados sin ayuda [ 22 ], nuestras observaciones subrayan la necesidad de complementar las instrucciones tradicionales en papel con otras herramientas educativas, como un video corto, que fue preferido por el 28,3% de los participantes del estudio para una mejor comprensión de las instrucciones de uso. Estos hallazgos recuerdan las recomendaciones anteriores de la OMS para la autoevaluación del VIH, de que todos los autoevaluadores deben tener la opción de acceder a la asistencia por teléfono, Internet o instrucciones adicionales como videos, animaciones o diagramas [ 41]. Debe enfatizarse la dificultad de explicar que el hisopo debe tocar la parte inferior de la pared interna de la fosa nasal para una correcta toma de muestras de NMT. Esta dificultad, encontrada en el 5,6% de los participantes, podría ser la causa de resultados falsos negativos.En el subestudio 2, todos los participantes del estudio llevaron a cabo el COVID-19 Ag-RDST y lograron obtener un resultado de prueba válido con un índice de usabilidad general estimado en 99,1%. El tiempo de realización de la prueba fue especialmente corto (8,1 min). Alguna dificultad en la correcta introducción del hisopo nasal en ambas fosas nasales y la necesidad de girar el hisopo 6 veces en el tubo de diluyente fueron las principales preocupaciones reportadas encontradas y fueron la razón más común para la ayuda oral. Todos los participantes que utilizaron las instrucciones en video realizaron la autoprueba fácilmente. Estas características subrayan la importancia de las instrucciones en video, cuando están disponibles. El uso de una línea directa también podría proporcionar asistencia remota directa.La capacidad de leer e interpretar correctamente los resultados de la autoevaluación se considera un paso delicado en la autoevaluación [ 42 ]. Esto se refiere no solo a la capacidad visual relacionada con una buena agudeza visual al leer e interpretar los resultados, sino también a la intensidad de las bandas a leer en la tira. En nuestra serie, la tasa (96,2%) de interpretación correcta de los resultados de COVID-19 Ag-RDST fue alta, como suele observarse, por ejemplo, con la autoprueba del VIH con un casete similar [ 28 , 29 , 30]. En un caso, los resultados de la prueba malinterpretados se referían a una banda positiva débil, leída como negativa. Esta dificultad para leer algunas bandas positivas débiles e interpretar definitivamente los resultados de la prueba puede ocurrir tanto en usuarios no profesionales como en usuarios capacitados en pruebas profesionales [ 43 ]. La lectura errónea de una tira débilmente positiva es obviamente restrictiva porque el sujeto cree falsamente que es negativo. Sin embargo, las instrucciones de uso establecen claramente que una prueba negativa no significa que el Ag-RDST no sea contagioso. La lectura errónea de pruebas no válidas interpretadas como positivas debe, en principio, corregirse mediante más pruebas moleculares de COVID-19, que deben ser sistemáticas en caso de positividad, como se indica en las instrucciones de uso.Las respuestas postest al cuestionario de satisfacción sobre las instrucciones de uso (subestudio 1), la realización del autodiagnóstico (subestudio 2) y la interpretación de los resultados (subestudio 3), mostraron que la gran La mayoría de los pasos de autoprueba de COVID-19 fueron considerados fáciles o muy fáciles por la mayoría de los participantes, como se informó anteriormente para la autoprueba de VIH utilizando un casete de prueba rápida similar [ 28 , 30 ]. Se agradeció especialmente ver un video antes de la prueba. La voluntad de repetir el uso del estudio Ag-RDST cuando sea necesario es importante para un uso generalizado, porque el Ag-RDT para la detección del SARS-CoV-2 debe repetirse para identificar individuos infecciosos, o para uso personal privado para que se convierta en una rutina. .En conjunto, nuestras observaciones destacan en adultos la usabilidad de la autoprueba de antígeno BIOSYNEX COVID-19 Ag + utilizando el auto muestreo indoloro de NMT como un enfoque novedoso para evaluar la infección por SARS-CoV-2 mediante el uso de la prueba Ag-RDT y la autointerpretación. de los resultados en un entorno supervisado. La viabilidad de la autocolección de hisopos nasales en la comunidad se enfatizó anteriormente para el diagnóstico de infección por virus respiratorios, como la influenza A y B y otros virus respiratorios comunes, incluidos los coronavirus comunes [ 44 , 45 ], y estos hisopos parecen ser ambos fácil de realizar [ 42 , 43 , 44 , 46 , 47 , 48 ] y bien aceptado [ 46, 48 ]. La autocolección de secreciones nasales para virus respiratorios ofrece beneficios potenciales significativos al reducir la necesidad de equipo de protección personal, limitar la exposición de los pacientes y el personal a infecciones, aumentar la comodidad y el acceso para los pacientes y la puntualidad de la recepción de la muestra [ 45 , 48 , 49 ] . En la presente serie, el manejo de la prueba en sí no supuso ningún problema particular, como se demostró anteriormente para otras autopruebas con casetes similares [ 28 , 29 , 30 , 50]. El auto muestreo de NMT fue aceptable y alcanzable en una variedad de niveles de educación, y la mayoría de los participantes prefirieron este método sobre la recolección de atención médica, probablemente debido a la capacidad de los pacientes para controlar el nivel de comodidad de la recolección nasal mejor que un capacitado. recolector puede, como se ha informado anteriormente para otros virus respiratorios [ 46 , 47 , 48 ]. Finalmente, la interpretación de las dos bandas en la tira del estudio Ag-RDST fue en general correcta, como se mostró anteriormente para las pruebas de diagnóstico rápido de forma comparable en usuarios legos de la población adulta general [ 28 , 29 ].
4.2. Rendimiento analítico de usuarios legos de la autoprueba de antígeno BIOSYNEX COVID-19 Ag +
El rendimiento analítico del estudio Ag-RDST se evaluó por referencia a rtRT-PCR multiplex para la detección de ARN del SARS-CoV-2 como estándar de oro en un entorno comunitario de la vida real. En esta evaluación, la sensibilidad de la autoprueba de antígeno BIOSYNEX COVID-19 Ag + fue menor entre las muestras de personas asintomáticas (83,3%) que entre las muestras de personas sintomáticas (93,8%). La especificidad (> 99,0%) fue alta en muestras de grupos asintomáticos y sintomáticos. La prevalencia de resultados positivos de la prueba de SARS-CoV-2 rtRT-PCR en esta población fue relativamente alta (20,7% en general; 7,9% para participantes asintomáticos y 53,3% para participantes sintomáticos). La alta carga viral de SARS-CoV-2 en las vías respiratorias superiores e inferiores, incluidos los sitios nasales [ 41 , 47 , 48, 51 ] y las técnicas moleculares sensibles pueden explicar la sensibilidad equivalente de la autocolección nasal a las muestras recolectadas para el cuidado de la salud en pacientes con COVID-19.El estudio Ag-RDST cumplió con las recomendaciones actuales de la OMS para un cribado Ag-RTD que indica que, como mínimo, las Ag-RDT deberían identificar correctamente más casos de los que perderían (sensibilidad ≥80%) y tendrían una especificidad muy alta ( ≥97-100%) [ 52 ]. Además, los rendimientos analíticos deben ser de un orden comparable, ya que los participantes en nuestro estudio de Ag-RDT se informaron previamente para algunos Ag-RDT en formato de inmunoensayo de flujo lateral [ 9 , 53 , 54 , 55 , 56 , 57 , 58 , 59 , 60 , 61], mientras que varios estudios han informado niveles de sensibilidad mucho más bajos que contrastan con una especificidad siempre alta [ 6 , 62 , 63 , 64 , 65 ]. Además, la autoprueba de antígeno COVID-19 Ag + de Ag-RDST BIOSYNEX cumplió con las recomendaciones actuales de la Haute Autorité de Santé (Saint-Denis, Francia) para un cribado de Ag-RTD, indicando que, como mínimo, las Ag-RDT necesitan identificar correctamente más casos de los que se perderían en pacientes sintomáticos (sensibilidad ≥80%) así como en individuos asintomáticos (sensibilidad ≥50%) y deben tener una especificidad muy alta (≥99%) [ 66 ].Nuestros resultados muestran claramente que los rendimientos analíticos del estudio Ag-RDST fueron mejores en el caso de una carga viral alta, es decir, en el caso de una excreción viral significativa, especialmente en individuos sintomáticos. Estas observaciones confirman el interés diferencial de Ag-RDT para la detección de antígenos del SARS-CoV-2 en función del nivel de carga viral de la muestra analizada, subrayado por varios autores [ 67 , 68 , 69 ] e internacionales [ 52 ] y recomendaciones nacionales [ 66 , 70 ]. Como se indica para Ag-RDT [ 52 , 66 , 70], Los Ag-RDST se pueden utilizar en el mejor de los casos para detectar individuos sintomáticos infectados con SARS-CoV-2 que padecen síntomas similares al COVID-19 con cargas virales elevadas, por lo que son individuos altamente contagiosos.Estudios previos hasta la fecha han analizado el rendimiento de muestras auto-recolectadas supervisadas y no observadas para la detección molecular del SARS-CoV-2 [ 45 , 71 , 72 , 73 , 74 , 75 , 76 , 77 ]. Por lo tanto, Tu y sus colegas compararon la detección del SARS-CoV-2 utilizando una variedad de tipos de hisopos recolectados por ellos mismos (bajo observación de un profesional de la salud) por 530 pacientes ambulatorios infantiles y adultos sintomáticos (o sus padres o tutores), con hisopos nasofaríngeos recolectados por el personal de atención médica. profesionales como el patrón oro de comparación [ 72]. De los hisopos nasofaríngeos recogidos por el profesional sanitario, 51 (9,6%) fueron positivos. Las muestras autorecogidas de cornete medio mostraron una alta sensibilidad del 96,2% (87,0% a 100%) [ 72 ]. McCulloch et al. Han demostrado que la recogida de torundas nasales medias en el hogar sin supervisión era comparable a la recogida de torundas nasofaríngeas recogidas por el médico para la detección molecular del SARS-CoV-2 en pacientes sintomáticos, en particular en aquellos con cargas virales más elevadas [ 71 ]. El alto rendimiento analítico de las muestras recolectadas por ellos mismos, incluidas las muestras de NMT, para la detección molecular de virus respiratorios comunes se informó de manera similar [ 44 , 45 , 46 , 47 , 48 , 78]. Un metanálisis de nueve estudios que compararon el auto muestreo y el muestreo de los trabajadores de la salud para las pruebas de influenza informó una sensibilidad combinada del 87% y una especificidad del 99% para la auto recopilación [ 79 ].Por último, se evaluó el rendimiento de las muestras autocolectadas supervisadas para la detección de SARS-CoV-2 utilizando un Ag-RDT de SARS-CoV-2 incluido en la lista de la OMS en voluntarios adultos en el hospital Charité, Universitätsmedizin Berlin, Alemania [ 12 , 13 , 14 , 15 ]. Por lo tanto, el muestreo nasal (incluido el auto muestreo) evaluado frente al muestreo nasofaríngeo condujo a un rendimiento comparable con el SARS-CoV-2 Ag-RDT [ 12 , 13 , 15 ]. Klein y sus colegas ampliaron estos estudios evaluando el rendimiento clínico de un hisopo NMT supervisado y recolectado por ellos mismos y un hisopo nasofaríngeo recolectado por un profesional, utilizando Panbio ™ Ag-RDT (distribuido por Abbott) en referencia a pruebas moleculares [ 14]. Los participantes pudieron realizar de forma fiable el automuestreo de NMT. Se diagnosticó una infección por SARS-CoV-2 mediante rtRT-PCR en 45 de 290 participantes (15,5%). Al comparar el NMT y el muestreo nasofaríngeo, el porcentaje de concordancia positiva del Ag-RDT fue del 88,1% (IC del 95%: 75,0-94,8%). La sensibilidad de Panbio ™ Ag-RDT estuvo entre 84,4% y 88,9%. La especificidad fue> 99,0% para NMT y muestreo nasofaríngeo. La sensibilidad de Panbio ™ Ag-RDT en participantes con alta carga viral (> 7 log10 copias de ARN del SARS-CoV-2 / mL) fue del 96,3% (IC del 95%: 81,7–99,8%) tanto para el NMT como para el muestreo nasofaríngeo. El automuestreo de NMT supervisado arrojó resultados comparables al muestreo de NP. En general, nuestros hallazgos y el estudio de Klein demuestran que el automuestreo de NMT conduce a resultados comparables al muestreo nasofaríngeo cuando se usa un Ag-RDT conveniente.13 ]. En el mismo entorno, Nikolai y sus colegas demostraron además que el muestreo nasal anterior profesional y NMT tenían una precisión equivalente para un SARS-CoV-2 Ag-RDT en adultos sintomáticos ambulatorios, pero no evaluaron el auto muestreo nasal anterior para Ag-RDST [ 13 ]. Curiosamente, los Centros para el Control y la Prevención de Enfermedades (Atlanta, GA, EE. UU.) Han agregado recientemente tanto el auto muestreo para el análisis en un laboratorio de referencia como el auto muestreo domiciliario de NMT como una alternativa aceptable al muestreo nasofaríngeo profesional en su guía para el SARS -Prueba de CoV-2 [ 11]. Autopruebas directas de COVID-19 y pruebas caseras que han recibido el estado de Autorización de uso de emergencia (EUA) de la Administración de Drogas y Alimentos de los EE. UU. (FDA) y están en el mercado, e incluso están disponibles en farmacias comunes en los EE. UU. La FDA también ha autorizado las autopruebas basadas en fluidos orales en virtud de una EUA, aunque se justifican más estudios clínicos sobre la sensibilidad de la saliva como material de muestra para la autoprueba de COVID-19.Otras tecnologías en desarrollo podrían utilizarse como autoprueba. Por ejemplo, la amplificación isotérmica mediada por bucle (LAMP) es un método de amplificación de ADN que permite la detección rápida y sensible de un gen específico. LAMP combinado con transcripción inversa (RT-LAMP) se ha utilizado con éxito para la detección de varios virus de ARN respiratorios, incluido el SARS-CoV-2 [ 80 ]. Recientemente, se ha desarrollado un dispositivo basado en papel de bajo costo y facilidad de uso para la detección sin extracción de SARS-CoV-2 en saliva completa usando RT-LAMP con una respuesta colorimétrica visible para el ojo humano, que podría usarse como una autocomprobación o un diagnóstico en el hogar debido a su simplicidad [ 81 ].
Conclusiones
Ag-RDST se puede utilizar para detectar individuos sintomáticos infectados con SARS-CoV-2 que padecen síntomas similares al COVID-19 con cargas virales elevadas y tiene el potencial de determinar individuos altamente contagiosos [ 52 , 66 , 70 ].
La detección a gran escala de poblaciones con infección activa por SARS-CoV-2 es una posible forma de romper las cadenas de transmisión para limitar la pandemia actual y desconfinar las sociedades. Si bien el cribado poblacional repetido de individuos asintomáticos se puede utilizar para limitar la propagación del SARS-CoV-2, la velocidad de notificación es mucho más importante que la sensibilidad, porque los resultados relacionados con el cribado de individuos infectados y asintomáticos siempre deben conocerse rápidamente [ 18].
Por lo tanto, a pesar de una menor sensibilidad para detectar infecciones, Ag-RDST para la detección del antígeno del SARS-CoV-2 puede ser una herramienta importante para el cribado debido a su rápido tiempo de respuesta y menores costos [ 67 , 68 ]. La rápida realización de Ag-RDST puede ayudar a limitar la transmisión del SARS-CoV-2 al identificar más rápidamente a las personas infecciosas a aislar, especialmente cuando se utiliza en el contexto de estrategias de pruebas en serie.La necesidad de servicios de salud ha crecido en todo el mundo durante la pandemia de COVID-19. Se requieren formas innovadoras de abordar esta crisis. La recolección automática de hisopos nasales para las pruebas antigénicas del SARS-CoV-2 ofrece una alternativa aceptable y confiable a las muestras recolectadas por los trabajadores de la salud. Por lo tanto, la recolección de hisopos asociados con Ag-RDST presenta varias ventajas, que son aún más importantes en un momento de crisis de salud global, incluida la accesibilidad fuera del sistema de atención médica, minimizando el uso de equipos de protección personal, limitando la exposición de los pacientes y el personal a infección y proporcionar una experiencia más cómoda para el paciente. La autocolección para pacientes infectados con SARS-CoV-2 o COVID-19 es bien aceptada, segura y escalable en el entorno pandémico.16 ].
Las altas sensibilidades de Ag-RDST, especialmente en pacientes sintomáticos, alivian las preocupaciones sobre el aumento de falsos negativos en este contexto. A medida que las sociedades se reabren, la expansión de las pruebas se vuelve fundamental para prevenir un resurgimiento global de COVID-19.
El auto muestreo nasal con Ag-RDST tiene el potencial de desempeñar un papel fundamental en el aumento del acceso a las pruebas en la población en general.
Actualización del informe del GTM sobre pruebas de diagnóstico de COVID-19
Actualmente las pruebas diagnósticas que existen se basan en la detección de: 1.- Material génico del virus 2.- Antígenos virales 3.- Anticuerpos generados frente al virus Las técnicas PCR y las de detección de material génico y antígenos virales, respectivamente, están muy bien establecidas, presentan una gran especificidad y sensibilidad, y permiten saber si hay presencia de estos elementos del virus en un momento concreto, aunque no indican en qué momento de la infección nos hallamos. Por otro lado, la generación de anticuerpos por parte del sistema inmunitario es una respuesta conocida que sigue una secuencia temporal y ayuda a identificar el momento del proceso infeccioso en que se encuentra el paciente. También, permite la identificación de individuos que han sido huésped del virus, inclusive cuando estos han sido asintomáticos durante la infección.
1.- DETECCIÓN DEL MATERIAL GÉNICO DEL VIRUS POR TÉCNICAS MOLECULARES 1.1 PCR La técnica de referencia para detectar la presencia del material génico específico del virus SARS-CoV-2 es la reacción en cadena de la polimerasa (PCR). La muestra utilizada suele ser un exudado nasofaríngeo aunque también puede utilizarse saliva, esputo, aspirado traqueal o lavado broncoalveolar. Actualmente, la PCR es la técnica de diagnóstico más sensible y específica de la que se dispone en esta pandemia (https://pubmed.ncbi.nlm.nih.gov/32570045/). La PCR detecta específicamente secuencias específicas del ARN del SARS-CoV-2, tanto genómico como en estado replicativo, desde la fase asintomática de la incubación hasta varias semanas después de la resolución del cuadro clínico. A pesar de su elevada sensibilidad, se han descrito porcentajes variables de falsos negativos debidos a factores asociados con la obtención y procesamiento de la muestra y con la dinámica de la carga viral. En cuanto a esta última, sabemos que es máxima dentro de los primeros 7 días de la infección, aunque algunas personas infectadas pueden retener material génico del virus hasta pasadas semanas e incluso meses, especialmente en pacientes que no resuelven el cuadro clínico. Por el contrario, la positividad que se detecta tras la resolución del cuadro clínico, especialmente en pacientes que desarrollan anticuerpos IgG neutralizantes frente al virus, se suele asociar a una infectividad mínima o nula. El porcentaje variable de falsos negativos se considera que es mayor en los primeros 5 días después de la infección (hasta de un 67%), y mínimo en el día 8 (21%), por lo que sería aconsejable que las pruebas PCR se realizaran 1–3 días después de la aparición de los síntomas para así minimizar los falsos negativos. Además, en pacientes asintomáticos, deberían tenerse en cuenta los ciclos de PCR a los cuales se considera que la muestra es infectiva. Las PCR positivas a ciclos (Ct) mayores de 35 tras un periodo de sintomatología leve o pasados los días preceptivos de cuarentena de incubación del virus, deberían de ser considerados despreciables en cuanto al significado clínico y los pacientes como no infecciosos. La dificultad de la interpretación de los resultados en algunos casos, junto con la complejidad de la obtención de la muestra, la laboriosidad de la técnica en sí, y la necesidad de llevarla a cabo en laboratorios especializados por técnicos expertos, son algunas de las limitaciones de la técnica PCR. 3 Existe una RT-PCR automatizada, muy rápida (30 minutos) y en funcionamiento como Point of Care (POC) (instrumento portátil), que utiliza el sistema Xpert Xpress (cepheid.com). Viene en un formato que no precisa de instalaciones especializadas ni de técnicos expertos. Está diseñada principalmente para situaciones de emergencia y necesidad de diagnóstico urgente y además se podría combinar con la detección de otros virus respiratorios (para filtrar falsos positivos). Con su uso se podría evitar la saturación de los servicios de urgencias. Así, se ha anunciado la combinación en un solo test de SARS-CoV-2, gripe A y B y virus respiratorio sincitial (Xpert Xpress SARS-CoV-2/Flu/RSV). Este equipamiento es muy específico, pero está disponible en la mayoría de los laboratorios de Microbiología para pruebas urgentes de detección de otros virus y ya se está empleando para SARS-CoV-2. 1.2 AMPLIFICACIÓN ISOTÉRMICA La amplificación isotérmica está basada también en técnicas moleculares y, en comparación con la PCR, requiere de una temperatura constante para la reacción de amplificación e identificación de un fragmento del material génico del virus. Existen numerosas variantes de esta técnica (https://pubmed.ncbi.nlm.nih.gov/32093592/). Este test es más rápido que la PCR (de 5 a 30 minutos). Se trata de una técnica muy sensible que puede tener, por tanto, las mismas dificultades de interpretación que se han comentado anteriormente para la PCR. Hay al menos cinco compañías que comercializan esta tecnología en forma de técnicas de Point of Care (instrumento portátil) que incluyen hisopo y kit de extracción y no precisan de técnicos expertos para su realización e interpretación. Una de estas técnicas es la empleada por el modelo comercial ID now de Abbott, que es uno de los sistemas de diagnóstico basados en esta técnica más extendidos en USA. (https://www.globalpointofcare.abbott/es/product-details/id-now.html) La amplificación isotérmica basada en bucle o LAMP (del inglés, Loop-mediated isothermal amplification) es una técnica específica de amplificación isotérmica casi tan sensible como la PCR. El pasado 17 de noviembre, la FDA aprobó el primer autotest para uso domiciliario denominado Lucira basado en esta técnica (https://www.lucirahealth.com/), aunque todavía no está disponible comercialmente y se necesitará prescripción médica para su uso. 1.3 CRISPR Este tipo de test, basado en el sistema CRISPR, detecta el material génico del virus mediante la hibridación de una sonda a una región concreta del material genético del virus. Esto permite, a su vez, la unión de una enzima Cas al ADN, que da como resultado la activación de una sonda que emite fluorescencia tras el procesamiento. Al depender de una actividad enzimática, la señal es 4 amplificable y acumulable, lo que permite una detección muy sensible del material génico del virus sin necesidad de su procesamiento. El test CRISPR requiere, al igual que las amplificaciones isotérmicas, de una mínima manipulación y se espera que pueda llegar a ser un test de autodiagnóstico en fechas no muy lejanas. Al igual que las anteriores, su alta sensibilidad puede plantear problemas diagnósticos cuando detecten positivos en personas que ya no secretan virus infectivos. Actualmente hay tan solo un kit aprobado que emplea esta tecnología. (Se incluye dentro de un listado de test aprobados; https://www.360dx.com/coronavirus-testtracker-launched-covid-19-tests) Sherlock Biosciences. Sherlock CRISPR SARS-CoV-2 kit (EUA 5/6/2020) EUA: Emergency Use Authorization 1.4 PCR Digital Para solventar el problema de la sensibilidad cuando las cargas virales son bajas, diversas compañías están comercializando una tecnología PCR de alta sensibilidad denominada PCR digital (dPCR), basada en una técnica muy sofisticada que emplea microfluídica y análisis a nivel de gota. Empresas como Gnomegen LLC, PreciGenome LLC y Bio-Rad la comercializan. 1.5 SECUENCIACIÓN DEL VIRUS Otra opción diagnóstica es la secuenciación directa del virus. Esta técnica requiere de equipamiento más sofisticado y se emplea fundamentalmente para identificar mutaciones en la secuencia del SARS-CoV-2. Tiene interés para el estudio de posibles casos de reinfecciones así como de la evolución de las cepas y su posible implicación en el desarrollo de la pandemia. Así, puede detectar mutaciones con repercusión clínica en términos de patogenicidad, evasión frente a los tratamientos basados en fármacos antivirales y/o anticuerpos neutralizantes o la inmunidad neutralizante completa frente al virus que se espera de las vacunas. Esta técnica de secuenciación es la que ha permitido la rápida identificación de las mutaciones detectadas en UK y en Sudáfrica, que tienen mayor capacidad de infección, pero de similar virulencia. (Ver Tablas 1A y 1B) 5 VENTAJAS E INCONVENIENTES DE LAS TÉCNICAS MOLECULARES Ventajas: En general, la prueba de RT-PCR es el pilar del diagnóstico de COVID-19, debido a su sensibilidad, especificidad y viabilidad en comparación con el cultivo viral. Para su interpretación es necesario considerar el momento de la prueba de PCR en relación con los síntomas, el límite de detección del ensayo y el tipo de muestra. Las próximas mejoras en la RT-PCR están dirigidas a una mayor facilidad de procesamiento, ahorro de materiales y tiempos de entrega de resultados más rápidos. Permite el uso de pools de ARN: se ha demostrado que la sensibilidad clínica se mantiene con pools de hasta 8 pacientes. Esta aproximación es útil en situaciones de falta de reactivos y/o número de muestras elevado, con el fin de reducir costes y acelerar los procesos de cribado, especialmente en momentos de baja incidencia. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7308776/ Inconvenientes:
Toma de muestra: la toma de muestra nasofaríngea con un hisopo es la recomendada y se puede combinar con la orofaríngea, para aumentar la sensibilidad. Esta toma de muestra es molesta para los pacientes, por lo que actualmente, se han desarrollado una prueba para la detección en saliva. En un estudio prospectivo, la muestra de saliva se asoció con una sensibilidad similar a la del hisopo nasofaríngeo en la detección del ARN del SARS-CoV-2. https://pubmed.ncbi.nlm.nih.gov/32857487/.
Temperatura de mantenimiento de la muestra: refrigerada a 4oC si se procesa antes de 72 horas; congelada a -70oC o temperaturas más bajas si se necesita mantener durante más tiempo.
Extracción de ARN viral: Este paso es un factor limitante porque cada contenedor de hisopo se debe inactivar (65oC, 30 minutos o con solución inactivadora), manipulando en campana de bioseguridad, y de forma individualizada. 2.- DETECCIÓN DE ANTÍGENOS VIRALES DE CORONAVIRUS SARS-COV-2 (PRUEBAS DE ANTÍGENO). Estos test se basan en la captura de antígenos específicos del virus mediante sus anticuerpos específicos. Los antígenos del SARS-CoV-2 son los antígenos N, S y el dominio RBD, siendo el antígeno N el más abundante en el virus). La técnica empleada para el test de antígeno suele ser la inmunocromatografía (lateral flow). Se trata de tiras reactivas donde se adsorben los anticuerpos específicos frente a los antígenos. Se han comercializado diversos kits para la detección de antígenos del SARS-CoV-2, la mayoría de ellos como test rápidos (resultados en 15-20 minutos) 6 Es importante destacar que estos test solo proporcionan una respuesta si/no a la presencia de la infección y no permiten conocer cuál es el contenido de la carga viral en una muestra. Además de la inmunocromatografía, existen test diagnósticos para detección de antígenos basados en otras tecnologías como inmunofluorescencia y biosensores electroquímicos y ópticos, y algunos de ellos requieren equipos específicos para su medición. La mayoría de los test se han desarrollado para muestras nasales y orofaríngeas, pero recientemente han aparecido otros que pueden emplear saliva. La Comisión Europea (CE) ha adoptado una recomendación sobre el uso de los test rápidos de antígenos, con una guía con fecha 18 de noviembre de 2020 en la que indica que los test de antígeno deben estar provistos del marcado CE y presentar al menos un 80% de sensibilidad y un 97% de especificidad. (Ver Tabla 2) Su sensibilidad se incrementa si se realiza en los 5 primeros días desde el inicio de los síntomas o dentro de 7 días tras una exposición confirmada con un caso COVID-19, ya que deben ser muestras con alta carga viral para que el test de antígeno sea capaz de detectarla. Hay pocos datos en personas asintomáticas ya que las empresas que han desarrollado estos test no las incluyen como población diana (Figura 1). Estos test deberían usarse para el diagnóstico rápido de la infección, particularmente en circunstancias de transmisión comunitaria alta. Hay que tener en cuenta que actualmente hay numerosos test de antígenos en el mercado, pero no todos tienen la misma sensibilidad y especificidad, por lo que es necesario conseguir información sobre sus validaciones antes de decantarse por unos u otros. VENTAJAS E INCONVENIENTES DE LOS TEST DE ANTÍGENO Ventajas:
Los test rápidos son sencillos, no requieren equipos sofisticados ni personal muy cualificado, pero sí deben ser llevados a cabo por personal entrenado.
Son más rápidos y baratos que la PCR
Permite el aislamiento más temprano de los casos positivos.
Puede permitir rápidos estudios masivos en situación de alta prevalencia y/ de alto riesgo (ejemplo, residencias), y repetirlos en personas con alto riesgo potencial. Inconvenientes:
La mayoría de ellos tienen una sensibilidad inferior a los test RT-PCR
No hay datos en personas asintomáticas 7
La ventana de positividad es más estrecha que la de RT-PCR (Figura 1)
Los resultados negativos en una población con alta prevalencia de infección, deberían confirmarse por RT-PCR o repetir el test de antígeno.
COMPARATIVA DE LOS TEST RÁPIDOS PARA DETECCIÓN DEL VIRUS El diagnóstico de infección por SARS-CoV-2 se realiza típicamente utilizando RTPCR estándar, que requiere transporte de la muestra a un laboratorio especializado así como personal, equipos y materiales especializados. Debido a esta complejidad, existe un gran interés por las pruebas moleculares y de antígeno rápidas. Las pruebas rápidas moleculares y de antígenos difieren de las RT-PCR estándar en varios aspectos: facilitan el acceso a las pruebas mediante ensayos en el lugar de atención, mejoran la eficiencia ya que reducen los pasos del proceso entre la recogida de la muestra y los resultados y algunas también anulan la necesidad de un laboratorio central y de equipo especializado. La muestra para las pruebas rápidas se obtiene con un hisopo nasal, que suele ser mejor tolerado que el hisopo nasofaríngeo. 8 Las pruebas rápidas pueden contribuir en última instancia a una mejor contención del SARS-CoV-2 mediante una detección más eficaz y el posterior aislamiento. En consecuencia, algunos hospitales están utilizando o considerando la posibilidad de utilizar pruebas rápidas como parte de sus algoritmos de prueba del SARS-CoV-2. https://www.cdc.gov/coronavirus/2019-ncov/lab/resources/antigen-testsguidelines.html#table2 La evaluación de las pruebas de antígeno para el diagnóstico de la COVID-19 demuestran una especificidad similar a las pruebas RT-PCR estándar, pero una sensibilidad inferior que las pruebas moleculares rápidas, que muestran una sensibilidad similar a las pruebas RT-PCR estándar https://www.finddx.org/covid19/dx-data/. Las pruebas rápidas de antígenos y moleculares pueden diferir en sus características de rendimiento, por lo que es difícil extrapolar la evaluación de una prueba rápida específica a otras. Hasta que no se disponga de datos más fiables sobre la utilización de estas pruebas en los entornos clínicos, la RT-PCR estándar seguirá siendo probablemente la técnica más fiable para diagnosticar la infección por el SARSCoV-2. Sin embargo, en determinados escenarios, una prueba rápida puede ser una prueba de detección inicial razonable; entre ellos se incluyen situaciones en las que es importante el aislamiento rápido, como en los servicios de Atención primaria y de urgencias o casos en los que una persona sintomática ha tenido un contacto conocido con alguien con COVID-19. Así pues, no se hacen recomendaciones a favor o en contra del uso de pruebas rápidas (es decir, tiempo de resultado ≤ 1 hora) frente a las pruebas estándar de ARN en individuos sintomáticos sospechosos de tener infección por SARS-CoV-2. Varios ensayos clínicos en curso tienen como objetivo evaluar las pruebas rápidas de COVID-19 (https://clinicaltrials.gov/ct2/show/NCT04460690). En el caso del kit de prueba Lucira COVID-19 All-In-One, comentado anteriormente, es una prueba de PCR rápida que proporciona resultados en 30 minutos. 3.- DETECCIÓN DE ANTICUERPOS FRENTE AL VIRUS En este caso se detectan los anticuerpos o inmunoglobulinas generados frente al virus en muestras de sangre, suero, plasma o saliva, principalmente. La sensibilidad y especificidad de estas pruebas depende, sobre todo, de cuatro factores:
El isotipo de anticuerpos detectado. Las pruebas pueden detectar inmunoglobulinas totales, isotipos concretos (IgM, IgG e IgA) o sus combinaciones (Figura 1). La sensibilidad de todos ellos es baja durante los primeros días de infección porque su generación (seroconversión) requiere aproximadamente una semana. Las sensibilidades óptimas se encuentran en torno a las 3-4 semanas y a partir de entonces, la cinética es altamente variable. La IgM sería la primera en aparecer y también en decaer, aunque 9 hay casos descritos en los que perdura varios meses. Esta cinética puede complicar la interpretación de los resultados sobre todo cuando se miden simultáneamente dos o más isotipos.
El antígeno contra el que se prueban las inmunoglobulinas. Uno de los más habituales es el N (nucleocápside o nucleoproteína) aunque también hay pruebas que utilizan mezclas de varios antígenos, incluyendo el S y RBD, o de proteínas totales del virus.
El perfil inmune de cada individuo. El tiempo necesario para la seroconversión, la clase de anticuerpos predominantes y el hecho de que se mantengan o decaigan los anticuerpos, depende del estado general de salud e inmunidad de cada persona, del curso de la enfermedad, de las posibles comorbilidades y de los tratamientos recibidos.
El método de detección: o Los test de flujo lateral o inmunocromatografía se realizan en minutos y requieren tan solo una gota de sangre; son solo cualitativos y no requieren instrumental adicional. o El ELISA es una técnica muy sensible, habitualmente estos test son semi-cuantitativos y requieren del uso de equipos especializados e instrumental específico. o Los ensayos de quimioluminiscencia (CLIA) son análogos al ELISA en sensibilidad y requieren también de equipos especializados que permiten además el procesamiento masivo de muestras. Los ensayos basados en CLIA ofrecen más del 98% de sensibilidad dentro de los 11-15 días posteriores al inicio de los síntomas, mientras que en los test rápidos puede bajar al 53% en el caso de la IgM y al 70% en el caso de la IgG. Otro aspecto a considerar es que los datos de sensibilidad y especificidad se calculan en base al desempeño de los test en individuos con síntomas y PCR confirmada y no son trasladables a estudios de cribado de seroprevalencia o inmunización en los que hay gran número de asintomáticos. (Ver Tabla 3) CONCLUSIONES DE LOS TEST DE ANTICUERPOS La utilidad de los test de anticuerpos como método diagnóstico es limitada, dado que su sensibilidad en la primera semana tras el contagio es baja, aunque pueden utilizarse como apoyo al diagnóstico en los casos positivos. Sin embargo son esenciales para los estudios de seroprevalencia e inmunización. 10 Cuanto más completo sea el repertorio de clases de anticuerpos y antígenos testados, más fiel será el reflejo de la inmunidad humoral de cada persona. Sin embargo, la ausencia o bajada de los niveles de anticuerpos no puede asimilarse con ausencia de inmunidad, puesto que se ha podido generar memoria inmunitaria con linfocitos T (celular) y B (humoral), que responderán mejor frente al mismo patógeno en futuras exposiciones.
La autoevaluación como herramienta invaluable en la lucha contra la pandemia de COVID-19
La identificación de casos de COVID-19 ha sido impulsada principalmente por pruebas de diagnóstico molecular.
Las pruebas de diagnóstico molecular tienen limitaciones y algunos países han empleado pruebas de detección rápida de antígenos (RADT) como una herramienta para identificar rápida y eficazmente los casos positivos en las poblaciones.
Nuestra encuesta realizada en Grecia y Chipre a 248 participantes mayores de 18 años identificó una fuerte preferencia por la autoprueba de saliva en el hogar.
Un enfoque de detección mediante el uso repetido de RADT, incluso mediante la autoevaluación, presenta claros beneficios para mantener la incidencia de COVID-19 en niveles más bajos.
Debería emprenderse activamente un seguimiento y una evaluación adecuados de la implementación de RADT mediante autopruebas con un enfoque en los usuarios finales.
Introducción
Desde el inicio de la pandemia de COVID-19, la identificación de casos mediante pruebas ha sido un pilar fundamental de la respuesta global, con un enfoque en las pruebas moleculares mediante transcriptasa inversa-PCR (PCR) para el diagnóstico. La PCR sigue siendo el estándar de oro en todos los países, 1 y la mayoría de los países también lo emplean con fines de diagnóstico y vigilancia. A pesar de que la PCR puede proporcionar la medición de la carga viral a través del valor umbral del ciclo, los resultados todavía se informan de forma rutinaria como positivos o negativos, tratando a todos los individuos infectados de la misma manera. 2 Además, aunque la PCR es invaluable en el diagnóstico de COVID-19 y podría ser útil para rastrear la dinámica epidemiológica durante un brote, 3tiene limitaciones que incluyen el costo, la infraestructura de laboratorio que normalmente se requiere para realizar la prueba y la entrega de resultados de manera oportuna.
Se ha reconocido que las pruebas de detección rápida de antígenos (RADT), en forma de ensayos de flujo lateral, tienen un gran potencial para abordar las limitaciones de la PCR, en particular para los países de ingresos bajos y medianos. 4 Se han desarrollado como pruebas de laboratorio que requieren equipo especializado y como pruebas en el lugar de atención con fácil lectura de resultados y realización. La Organización Mundial de la Salud 5 ha publicado una guía sobre el uso de RADT y las recomienda para que las administren los profesionales de la salud, mientras que el Centro Europeo para el Control y la Prevención de Enfermedades (ECDC) publicó recientemente 2 informes técnicos que destacan las consideraciones sobre el uso de autoevaluaciones. con RADT tanto para personas asintomáticas como en entornos ocupacionales. 6 , 7La autoevaluación se define como el proceso mediante el cual una persona toma su propia muestra de la nariz / garganta (hisopo nasal, hisopado faríngeo, saliva o una combinación de los anteriores) y procede a realizar la prueba e interpretar los resultados por sí misma. Larremore et al 8, basándose en su modelo, han abogado por repensar las estrategias de salud pública actuales en las pruebas cambiando hacia la ampliación de las RADT, con un enfoque en garantizar el uso repetido y la frecuencia en todos los entornos y poblaciones en un esfuerzo por permitir que los países abrir sus sociedades y economías.
Desde una perspectiva de salud pública, las RADT son extremadamente útiles para identificar infecciones activas. 9 , 10 sino que también aumentan la accesibilidad de los individuos a las pruebas y gracias a la velocidad de obtención del resultado permitir la detección temprana de casos positivos, controlando además la transmisión de enfermedades. Además, la disponibilidad de RADT no solo ha mejorado la conveniencia para muchas personas que habrían tenido dificultades para llegar a un sitio de prueba, sino que también ha hecho que las pruebas sean más accesibles para aquellos que son vulnerables, se protegen, se autoaislan o esperan una cirugía hospitalaria electiva. 11Por otro lado, los RADT se basan en la voluntad y la capacidad del individuo para realizar correctamente la autocomprobación y reportar un resultado positivo, lo que lleva tanto a un subregistro como a un aumento en el número de falsos positivos y falsos negativos, según el panorama epidemiológico. 6 Por lo tanto, su mal uso podría dificultar el seguimiento de las tendencias de la enfermedad a lo largo del tiempo. Además, las muestras de autoevaluación no están disponibles para secuenciar y monitorear variantes de interés. 6
La renuencia de algunos países a probar métodos de detección y diagnóstico adicionales distintos de la PCR ha cambiado su enfoque hacia los grandes programas de vacunación. En un esfuerzo científico hercúleo, las vacunas específicas de dosis única o doble para COVID-19 (Pfizer, Astra Zeneca, Moderna, Johnson & Johnson) han recibido autorización de uso de emergencia por parte de las autoridades reguladoras y actualmente se están implementando en todo el mundo. La seguridad y los altos niveles de eficacia en la prevención de enfermedades sintomáticas demostrados en ensayos clínicos aleatorios se están documentando a partir de estudios de implementación de «eficacia» en la vida real en los EE. UU., Reino Unido e Israel. 12– 14 Los datos más recientes también han destacado el papel de las vacunas, incluidas las de Pfizer-BionTech y Moderna, en la prevención de infecciones asintomáticas en los vacunados, 12 aunque se ha informado de una disminución de la inmunidad esterilizante durante la circulación de la variante Delta. 15 A pesar de la gran promesa de reducir las hospitalizaciones y la mortalidad, existen cantidades limitadas de vacuna a nivel mundial, lo que está obstaculizando nuestros esfuerzos en la carrera por vacunar al mundo. 16 La vacilación ante las vacunas también está desafiando a los gobiernos en sus esfuerzos por lograr que las personas acepten la vacunación en un momento de mayor ansiedad y fatiga pandémica. 17– 20
En Europa, la autoevaluación se ha empleado como una herramienta complementaria en la identificación de casos y la identificación de personas que han desarrollado alguna forma de inmunidad al SARS-CoV-2, lo que permite a las personas regresar al trabajo de manera segura y obtener información sobre la evolución. de la epidemia, incluso cuando se ha alcanzado un umbral de inmunidad colectiva. 21– 24 gobiernos tanto en el Reino Unido como en Grecia han adoptado el uso de pruebas de sobretensión con RADT en un intento de abrir lentamente negocios, escuelas y tiendas minoristas y, más recientemente, para controlar los brotes locales de variantes del SARS-CoV-2, como el delta. -variante (B1.617.2) en puntos de acceso del Reino Unido. 25 El uso de RADT gratuitos para la detección del SARS-CoV-2 mediante hisopos nasofaríngeos se ofreció a todas las empresas en el Reino Unido en marzo de 2021, seguido de pruebas dos veces por semana para todas las personas asintomáticas a principios de abril de 2021. 24 De manera similar, Grecia ha introducido el uso de 2 RADT nasales gratuitos para uso semanal por personas asintomáticas en escuelas, empresas y entornos comunitarios. 26Aunque, el uso de pruebas de antígeno de flujo lateral fue autorizado por la Administración de Drogas y Alimentos de los Estados Unidos (FDA) 27 en marzo de 2020, inicialmente se requirió una receta para su uso por parte del público en general. Dos pruebas rápidas de antígenos caseras ahora se venden sin receta en los estantes de las farmacias, sin necesidad de receta médica para personas asintomáticas en los EE. UU., Lo que permite al país mitigar la cadena de transmisión y reducir la prevalencia del SARS-CoV-2. 28
A medida que más países se están moviendo hacia diferentes modos de prueba de COVID-19 al expandir sus políticas de prueba, estábamos interesados en investigar la aceptabilidad y viabilidad de las autopruebas. Para ello, realizamos una encuesta transversal de residentes en Grecia y Chipre mayores de 18 años, con un enfoque en la isla de Lesbos, Grecia en el período del 16 de enero al 16 de marzo de 2021 utilizando la plataforma en línea JISC, con el fin de determinar las preferencias de los participantes sobre las pruebas COVID-19 e identificar cualquier relación entre los datos demográficos particulares y sus puntos de vista sobre las autoevaluaciones.
Métodos
Diseño del estudio y participantes
La encuesta en línea se creó en la plataforma JISC, que está diseñada para instituciones educativas y de investigación ( https://www.onlinesurveys.ac.uk/about/ ). La encuesta se distribuyó a 1000 personas a través de listas de correo electrónico y redes sociales utilizando la URL pública https://uow-survey.onlinesurveys.ac.uk/covid-19-testing-and-long-term-symptoms y estuvo abierta en el período del 16 de enero al 16 de marzo de 2021. Todas las respuestas fueron anónimas y el comité de ética FSE de la Universidad de Wolverhampton proporcionó la validación de las preguntas de la encuesta y la aprobación ética (LSEC / 202021 / PG / 52).
Las personas mayores de 18 años fueron reclutadas a través de la red de gimnasios Les Mills, Grecia y la red académica y profesional de Paraskevi Goggolidou en Grecia y Chipre. Se empleó un muestreo estratificado para garantizar una distribución adecuada por sexo y edad de la encuesta en línea. Se requirieron diez o más respuestas por grupo de edad (18-24, 35-44, 45-54, 55-64, 65+) para que se incluyera en el análisis del estudio. Se hizo hincapié en la contratación de participantes en las personas que residen permanentemente en Lesbos, Grecia, debido a su ubicación geográfica y entorno representativo. Lesbos es la tercera isla más grande de Grecia, tiene un tamaño de población manejable y es representativa de la demografía griega. También tiene un gran centro de integración de refugiados y tuvo una fuerte pendiente en el número de casos positivos de COVID-19 a fines de 2020 / principios de 2021.
La encuesta estuvo abierta a los participantes durante la duración del estudio, pero el mismo titular de la cuenta de correo electrónico no pudo volver a acceder a ella una vez completada. La encuesta en línea consistió en la hoja de información del participante y el consentimiento, seguida de una serie de preguntas que comprenden: datos demográficos individuales y la capacidad del respondedor para emplear intervenciones no farmacéuticas, como el distanciamiento físico durante la pandemia de COVID-19; antecedentes de pruebas anteriores de COVID-19 y manifestación de la enfermedad; preferencia en el muestreo de COVID-19, la metodología de prueba y los entornos. Se proporciona un mapa de la encuesta en la Figura complementaria 1 . En todos los casos, cuando faltaban datos, se excluyeron las respuestas de este individuo en particular a todas las preguntas, de modo que solo se tuvieron en cuenta las respuestas completas.
Análisis de los datos
Como la medición de resultado utilizó datos categóricos recopilados, se utilizaron frecuencias para presentar datos descriptivos. Se realizaron pruebas de chi-cuadrado para comparar datos sociodemográficos y de actitud entre los grupos que estaban dispuestos a autoevaluarse y los que no querían / no saben y para los predictores demográficos de las preferencias de las pruebas. Se realizó un análisis de regresión logística para evaluar los factores que pueden predecir la voluntad de autoevaluarse. Se consideró estadísticamente significativo un valor de p <0,05.
Resultados
De las 1000 personas a las que se distribuyó la encuesta, 860 participantes accedieron a la encuesta en línea y se obtuvo una tasa de respuesta del 62%. Excluyendo las respuestas incompletas, se recibió un total de 248 respuestas completas. Alrededor del 60% de los que respondieron se basaron en la isla de Lesbos, Grecia, el 20% en el resto de Grecia y el 20% en Chipre. Las respuestas aceptadas fueron de distribución equilibrada por género (55% mujeres, 45% hombres) y características demográficas particulares de la región (97,6% blancos, 98% griego como lengua materna, 72% con estudios superiores, 83,1% cristianos, 62% empleados; tabla 1). Aproximadamente el 14% de los encuestados había sospechado o confirmado COVID-19 antes de participar en la encuesta y el 63% ya se había sometido a una prueba de diagnóstico de COVID-19 realizada por un profesional. Aproximadamente el 62% informó que no había podido mantener el distanciamiento social, el 59% trabajaba en espacios confinados y el 26% era un contacto cercano de un caso de Covid-19 (datos no mostrados).
Tabla 1. Resumen de la demografía de los participantes.
La mayoría de los participantes (79%; n = 196) informaron estar dispuestos a autoevaluarse y los individuos restantes informaron que no (10,5%; n = 26) o no saben (10,5%; n = 26). Para fines de análisis de datos, los grupos de no saber y no saber se combinaron (21%; n = 52) en un grupo (grupo de no saber / no saber). Se realizó una prueba de chi-cuadrado de Pearson para medir si había diferencias demográficas significativas entre los grupos que querían y que no o no sabían. El análisis reveló que aquellos que están dispuestos a autoevaluarse tienen más probabilidades de ser graduados universitarios que aquellos que estaban en los grupos no sabe / no sabe (χ 2 = 15,398, gl = 1, p <0,001). No se encontraron otras diferencias demográficas entre los grupos que quisieron y no / no saben ( Tabla 2 , resultados solo para las variables significativas).
Tabla 2. Un resumen de las diferencias significativas entre los grupos que estaban dispuestos a realizar una autoprueba de COVID-19 en casa y los que respondieron No / No sé.
Tabla 2. Un resumen de las diferencias significativas entre los grupos que estaban dispuestos a realizar una autoprueba de COVID-19 en casa y los que respondieron No / No sé.Ver versión más grande
Se realizaron modelos de regresión logística sobre variables de chi-cuadrado significativas para medir los predictores de la voluntad de autoevaluarse. Ser un graduado universitario predijo significativamente la probabilidad de estar dispuesto a autoevaluarse (razón de probabilidades [OR] = 3.455, P <.001). La ubicación del sitio de prueba no predijo la voluntad de autocomprobarse. Cuando se les preguntó sobre la preferencia por el método de muestreo, la mayoría de los participantes indicaron que preferían una prueba de saliva (73%; n = 180), seguida de una prueba de punción digital (13%; n = 32); hisopo nasal (11%; n = 27); o frotis de garganta (7%; n = 9). La prueba de chi-cuadrado de Pearson encontró diferencias significativas entre graduados universitarios y no graduados en el tipo de prueba COVID-19 preferida (χ 2 = 8.95, gl = 3, P <0,03); los graduados eran más propensos a preferir la prueba de saliva y menos propensos a preferir la prueba de punción en el dedo que los no graduados ( Tabla 3 ). Además, chi-cuadrado encontró que los no griegos eran significativamente más propensos a preferir la prueba de saliva y menos propensos a preferir el hisopo nasal que los griegos (χ 2 = 9.12, gl = 3, P <.028); no se detectaron otras diferencias significativas relacionadas con otras características de los participantes (por ejemplo, edad, sexo, idioma, empleo). Además, no se revelaron diferencias significativas entre las preferencias del método de muestreo de los individuos y su disposición a autocomprobarse o no / no sabe (χ 2 = 3.652, gl = 3, P > .05).
Tabla 3. La prueba de chi-cuadrado de Pearson encontró diferencias significativas entre graduados universitarios versus no graduados y griegos versus no griegos en el tipo de prueba COVID-19 preferida.
Tabla 3. La prueba de chi-cuadrado de Pearson encontró diferencias significativas entre graduados universitarios versus no graduados y griegos versus no griegos en el tipo de prueba COVID-19 preferida.Ver versión más grande
En particular, la mayoría de los participantes preferiría realizar la prueba una vez a la semana (40%; n = 100), <1 de cada 5 de las personas encuestadas optaron por realizarse la prueba una vez al mes (19%; 46), mientras que alrededor de 1 de cada 3 respondió que nunca deseaba hacerse la prueba (31%; n = 76). La prueba de Chi-cuadrado de Pearson encontró diferencias significativas entre los 2 grupos; los que estaban dispuestos a autoevaluarse prefirieron una vez a la semana, mientras que los que informaron que no / no saben prefirieron nunca ( Tabla 2 ). Por último, más de la mitad de los participantes preferirían realizar la prueba en casa (52%; n = 129) o no tenían preferencia en el lugar de la prueba (22%; n = 54). La prueba de chi-cuadrado de Pearson encontró diferencias significativas entre los 2 grupos (χ 2 = 36,331, gl = 6, P <0,001); el grupo dispuesto prefirió hacerse la prueba en casa, mientras que el grupo no / no sabe prefirió el hogar, la clínica privada o los entornos EODY / YDY ( Tabla 2 ).
Discusión
Nuestros datos muestran una preferencia general por la autoprueba de COVID-19 de la manera menos invasiva, con una preferencia por la saliva como material biológico de elección. En las prácticas actuales de autoevaluación, generalmente se adquieren hisopos nasales. 29 La saliva debe considerarse como una muestra biológica adicional para la prueba de COVID-19, ya que aunque su sensibilidad es menor que la de la PCR con hisopo, 30 es un material clínicamente aceptable 31y su facilidad de uso permite realizar pruebas repetidas y sencillas, lo que aumenta la probabilidad de detección de casos positivos. Además, la naturaleza no invasiva de la adquisición de saliva permite la recolección de muestras en niños, personas discapacitadas, vulnerables o ansiosas y donde los recursos son escasos y, aunque tiene limitaciones, puede aumentar la aceptación de pruebas repetidas. 29
El concepto de autoevaluación era muy novedoso en el momento en que diseñamos nuestra encuesta. Aunque nuestro estudio fue de naturaleza transversal, la población de la encuesta fue limitada y se observó una sobrerrepresentación de graduados universitarios en la población de la encuesta, nuestros hallazgos nos dan una idea de la preferencia de los individuos por la autoevaluación. La encuesta se realizó en una población adulta que representa la demografía del país, siendo la única barrera para la participación el acceso a un dispositivo tecnológico. Esto se abordó con la ayuda de voluntarios locales que ayudaron a 7 participantes sin acceso en línea a completar los cuestionarios. Algunos participantes también presentaron inicialmente respuestas incompletas; donde esto era evidente, nuestros voluntarios locales los ayudaron a volver a enviar una respuesta completa.32 y, como tal, estos hallazgos solo reflejan la población principalmente griega. En el futuro, esta limitación podría abordarse diversificando los puntos desde los cuales los participantes accedieron a la encuesta, ya que esto podría haber mejorado la diversidad de la población de la muestra. Otras limitaciones del estudio incluyen el hecho de que se completó antes de que se lanzara en Grecia la autoprueba de detección asintomática; Es posible que después de la exposición repetida y la realización de pruebas nasales dos veces por semana, las preferencias de los participantes sobre la frecuencia y el método de prueba hayan cambiado. Nuestro estudio no accedió a las preferencias de las personas menores de 18 años, por lo que no se puede sacar ninguna conclusión sobre sus puntos de vista sobre la autoevaluación.
El uso de RADT de autoevaluación podría representar una parte vital para ayudar a un gobierno a levantar con cautela las restricciones para abrir su economía y sociedad. 6 , 7 , 30 También puede ser crítico durante un rápido aumento de casos, como se demostró más recientemente en la India, donde la guía del gobierno indica que una prueba casera de antígeno reactivo es un positivo definitivo. 33 Al 31 de marzo de 2021, la Administración de Alimentos y Medicamentos de los EE. UU. (FDA) había autorizado 4 pruebas para el uso de venta libre sin receta para pruebas seriadas asintomáticas para el SARS-CoV-2. 34 En Grecia, los RADT de autoevaluación se están distribuyendo a todos los ciudadanos registrados a través de farmacias desde principios de abril 35y en el Reino Unido se llevó a cabo un enfoque similar para la autocomprobación dos veces por semana. 24 El uso de un sistema en línea para informar resultados reactivos de COVID-19 en un ensayo de flujo lateral es esencial y el NHS lo ha aplicado con éxito mediante la aplicación Trace and Contact en el Reino Unido. 36 Las personas con un resultado reactivo para COVID-19 en Grecia lo informan a través de una plataforma en línea. 37 Además de las pruebas, el rastreo de contactos debe complementar la identificación de casos, ya que permite romper las cadenas de transmisión mediante la identificación rápida de conglomerados o brotes en entornos específicos, 38 e incluso si los casos positivos no informan formalmente o informan erróneamente su resultado, pueden informar a sus contactos, lo que puede tener un impacto positivo en el control de la transmisión.
Por lo tanto, aunque la autocomprobación puede mejorar la respuesta de un país contra COVID-19 incluso a medida que aumenta la cobertura de vacunación, es necesario considerar una serie de factores antes de que la autocomprobación pueda ampliarse de manera universal y confiable. En Europa, los fabricantes deben demostrar el cumplimiento de los requisitos legales aplicables de la Directiva de la UE 98/79 / EC para dispositivos médicos de diagnóstico in vitro, a fin de poder comercializar un RADT. 39 Ya se han expresado preocupaciones sobre la alta tasa de falsos negativos en RADT con sensibilidades del 72% y 58% observadas en cohortes de participantes sintomáticos y asintomáticos, respectivamente. Además, una revisión Cochrane publicada recientemente informó variaciones en las sensibilidades entre las marcas que van desde el 34% al 88% 40y otro estudio investigó RADT con características de rendimiento prometedoras, identificando Innova RADT como una de esas pruebas con excelente especificidad. 11 A medida que el mercado de kits de detección de venta libre se expande rápidamente a través de programas de autoevaluación respaldados por el gobierno, la capacidad de registrar y evaluar de manera centralizada la eficacia de diferentes pruebas de antígenos se vuelve primordial.
Para que los enfoques de autoprueba masiva tengan éxito en romper las cadenas de transmisión, los países deberán centrarse en los usuarios finales, proporcionar las pruebas de manera amplia y libre y generar confianza en el proceso. Nuestros hallazgos muestran que el 52% de los participantes prefieren hacerse la prueba en casa en comparación con otros entornos alternativos y el 31% se muestra reacio a hacerse la prueba debido al costo financiero. La autoevaluación de fácil acceso y libre disponibilidad sería una estrategia para abordar estas barreras. Para poder obtener resultados significativos, los usuarios deberán recibir instrucciones claras sobre cómo realizar las autoevaluaciones y algunos miembros clave de la comunidad deberán recibir capacitación y actuar como embajadores de las autoevaluaciones. Además, será necesario emplear un enfoque proactivo al informar un resultado, ya sea con la adopción de aplicaciones especialmente diseñadas o mediante un sitio web dedicado y de fácil acceso. En Grecia, desde que se puso en marcha la autocomprobación gratuita, se distribuyeron 42 millones de RADT a 4,6 millones de ciudadanos y se confirmaron unos 85 000 casos positivos mediante PCR.41 Los diferentes países tienen diversas prácticas sobre la validación de los resultados de las autopruebas y nuestra recomendación es que un resultado reactivo deberá ser validado por PCR, de una manera que no perjudique ni cause daño al individuo o la sociedad; El auto muestreo y la recolección casera de muestras para validación podrían ser una solución. También hay que señalar que, si bien el número de falsos positivos variará dependiendo de la prevalencia de base, 8 , 9 el éxito de este enfoque dependerá de la participación de la población que puede ser estimulado por las campañas de educación y embajadores de la comunidad, la efectiva y oportuna notificación y validación y la prestación de apoyo estatal a los casos positivos durante el período de cuarentena y aislamiento.
Conclusiones
Aunque la inmunidad inducida por vacunas es el vehículo para controlar y poner fin a la pandemia, la autoprueba de COVID-19 como parte de un enfoque de prueba integral debe priorizarse en paralelo a medida que avanzan las campañas de vacunación en todos los países. Se debe realizar un seguimiento y una evaluación adecuados de la implementación de la RADT mediante autopruebas, donde las métricas de las pruebas se comparten rápida y públicamente, como se ha hecho con la implementación de la vacunación en los países. Es importante destacar que la autoprueba con RADT debe entenderse como una estrategia de detección, en lugar de una prueba de diagnóstico, mientras que la saliva debe considerarse como un material de muestreo adicional, especialmente en casos de poblaciones jóvenes, vulnerables y de difícil acceso. Un enfoque de detección mediante el uso repetido de RADT, incluso mediante autoevaluación, presenta claros beneficios sobre lo que se considera una menor sensibilidad analítica del ensayo en comparación con las pruebas de diagnóstico molecular más costosas. Las pandemias requieren innovación y audacia en el desarrollo de políticas. Ampliar las pruebas para incluir la autoprueba puede ser fundamental para mantener la incidencia de COVID-19 en niveles más bajos, especialmente a medida que avanzamos hacia la siguiente fase de la respuesta global de COVID-19 en una era de cobertura de vacunación creciente, aunque limitada.
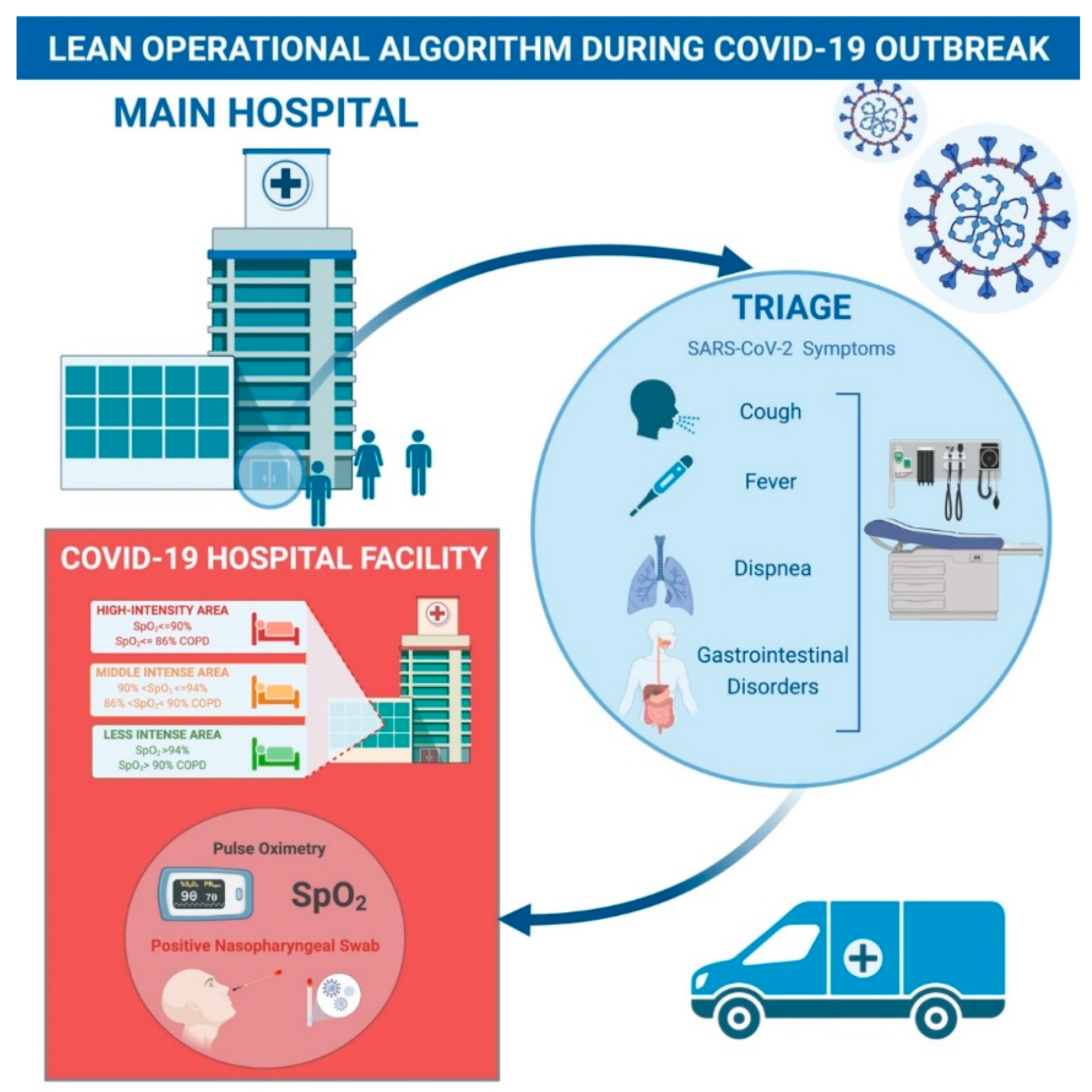

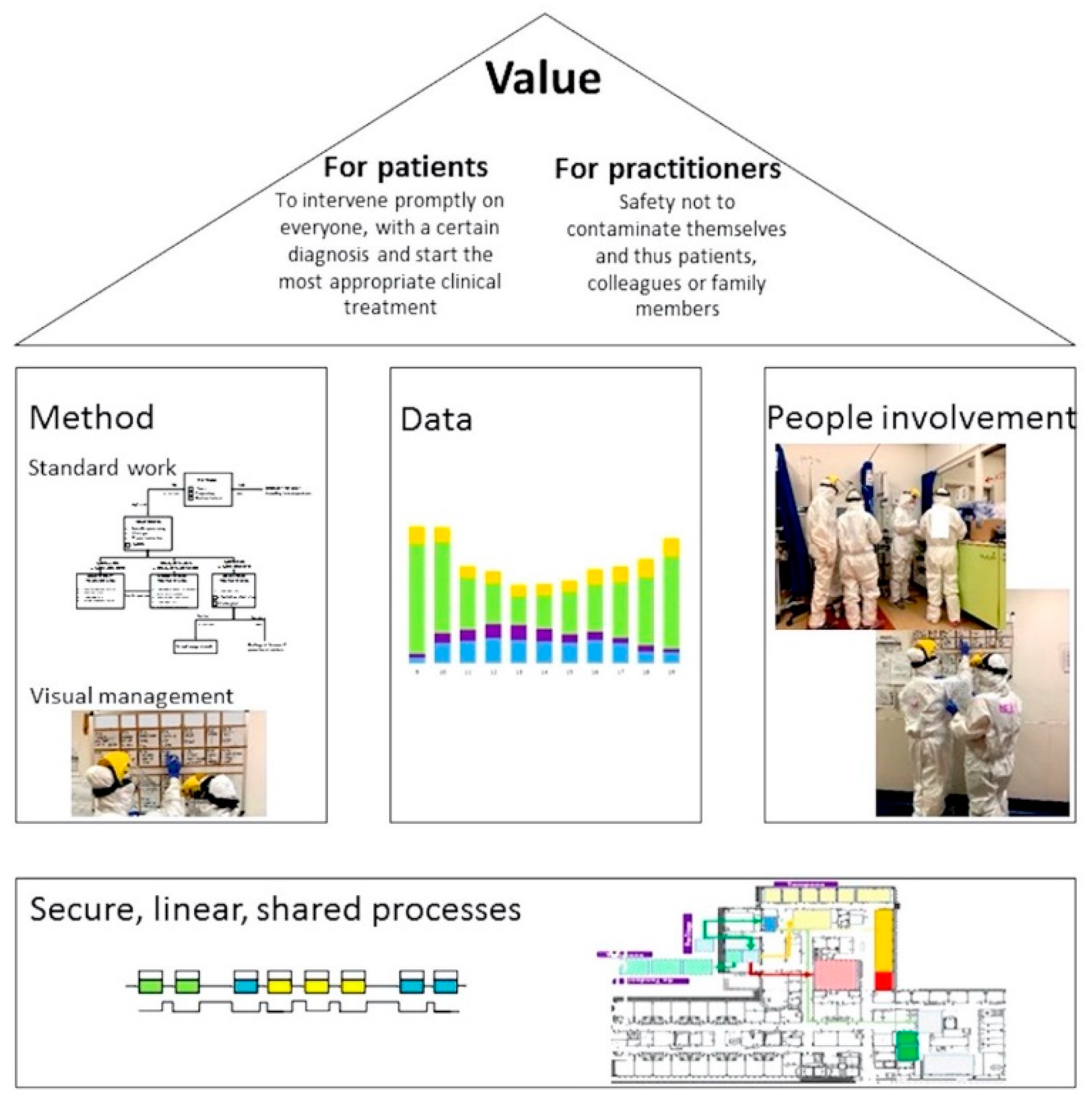









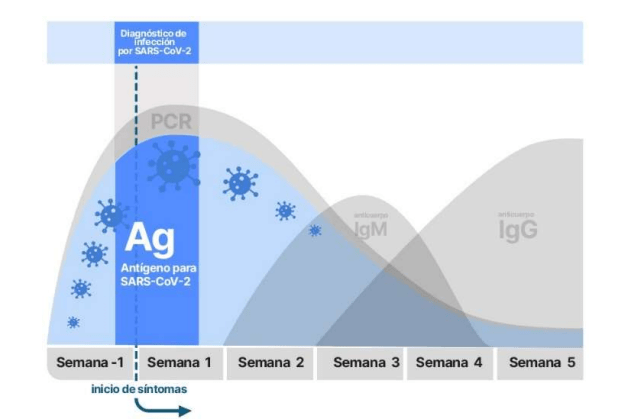




00409-4&id=gr1.gif){kind=link}
00409-4&id=gr2.jpg){kind=link}
00409-4&id=gr3.jpg){kind=link}
00409-4&id=gr4.jpg){kind=link}